Prasugrel
Biên soạn và Hiệu đính
Dược sĩ Xuân Hạo
Danh pháp
Tên chung quốc tế
Tên danh pháp theo IUPAC
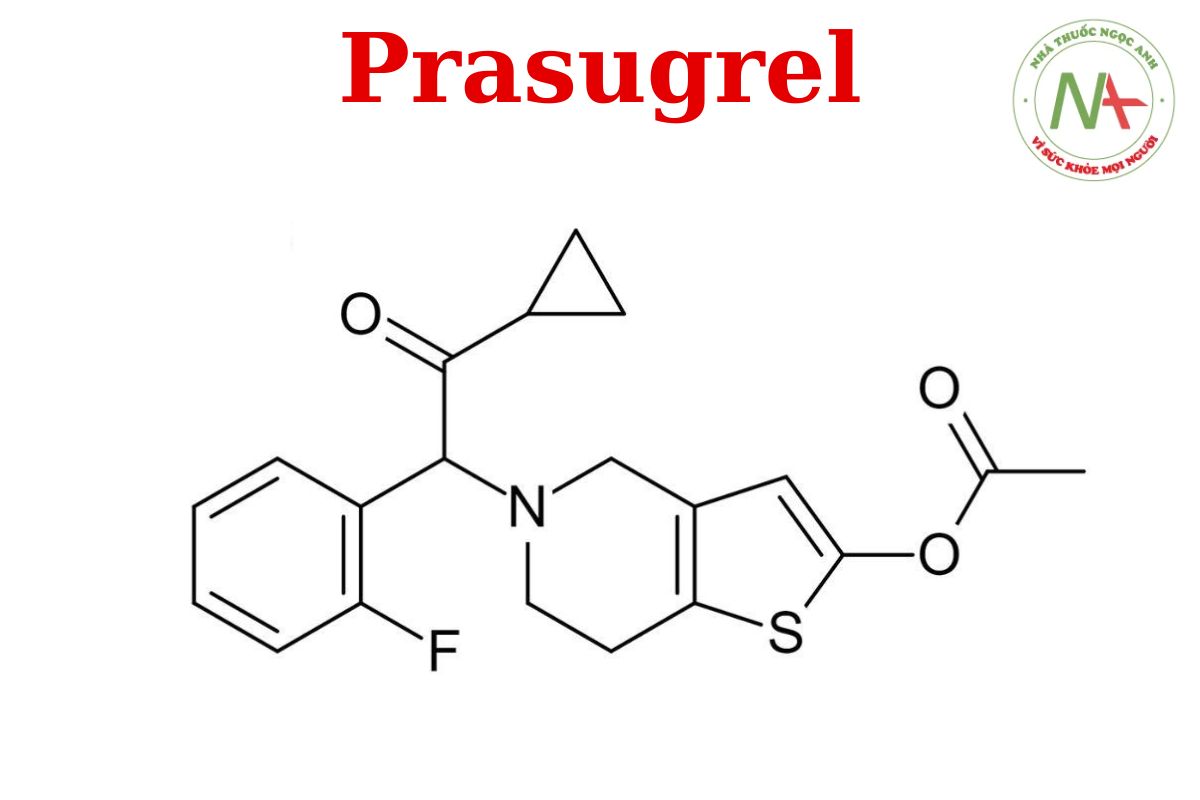
[5-[2-xyclopropyl-1-(2-fluorophenyl)-2-oxoetyl]-6,7-dihydro-4 H -thieno[3,2-c]pyridin-2-yl] axetat
Nhóm thuốc
Thuốc chống kết tập tiểu cầu
Mã ATC
B – Máu và cơ quan tạo máu
B01 – Thuốc chống huyết khối
B01A – Thuốc chống huyết khối
B01AC – Thuốc ức chế kết tập tiểu cầu, trừ heparin
B01AC22 – Prasugrel
Phân loại nguy cơ cho phụ nữ có thai
B
Mã UNII
34K66TBT99
Mã CAS
150322-43-3
Cấu trúc phân tử
Công thức phân tử
C20H20FNO3S
Phân tử lượng
373,4
Cấu trúc phân tử
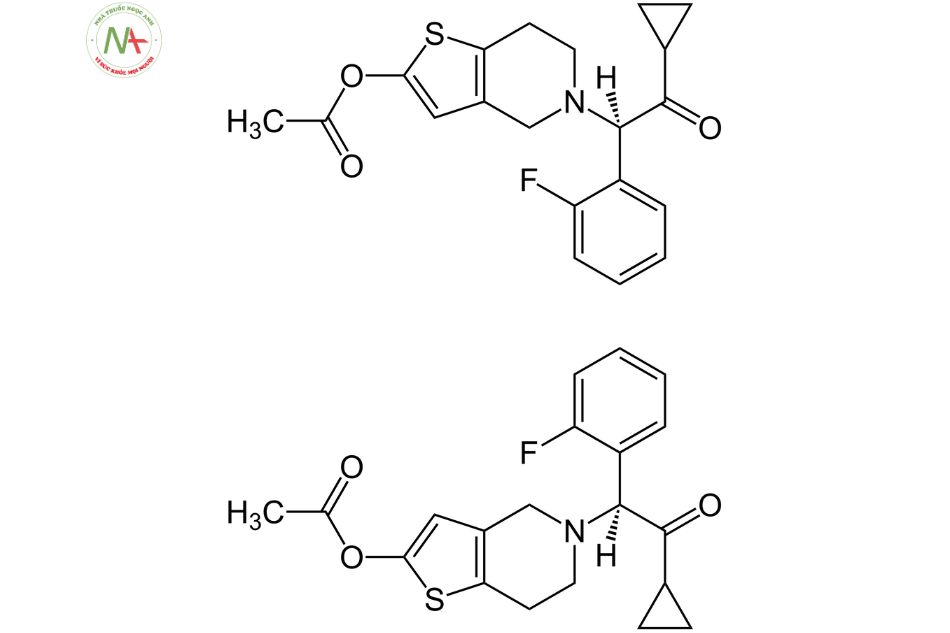
Prasugrel là một dẫn xuất thienopyridine
Các tính chất phân tử
Số liên kết hydro cho: 0
Số liên kết hydro nhận: 6
Số liên kết có thể xoay: 6
Diện tích bề mặt tôpô: 74,8 Ų
Số lượng nguyên tử nặng: 26
Các tính chất đặc trưng
Điểm nóng chảy: 189.04 °C
Điểm sôi: 449.57 °C
Độ tan trong nước: 0,00237 mg/mL
Hằng số phân ly pKa: 5.12
Chu kì bán hủy: 7.4 giờ
Khả năng liên kết với Protein huyết tương: 98%
Cảm quan
Prasugrel có dạng bột kết tinh màu trắng, tan được trong nước, metanol và ít tan trong 1- và 2-propanol và acetone.
Dạng bào chế
Viên nén: 5 mg; 10 mg.
Độ ổn định và điều kiện bảo quản
Prasugrel nên được bảo quản trong bao bì gốc của nhà sản xuất, ở nhiệt độ từ 15 đến 30 °C và tránh ánh sáng trực tiếp.
Nguồn gốc
Prasugrel được phát triển bởi Công ty Daiichi Sankyo và được FDA chấp thuận vào năm 2009.
Dược lý và cơ chế hoạt động
Prasugrel là một thienopyridin và là tiền chất ức chế thụ thể ADP bằng cách tác động không thuận nghịch lên thụ thể P2Y12 trên tiểu cầu. Chất chuyển hóa có hoạt tính của prasugrel ngăn chặn sự gắn kết của adenosine diphosphate (ADP) với thụ thể tiểu cầu của nó, làm suy yếu hoạt hóa qua trung gian ADP của phức hợp glycoprotein GPIIb/IIIa, dẫn đến ức chế kết tập tiểu cầu. Prasugrel được cho là có cơ chế tác dụng tương tự như clopidogrel.
Ứng dụng trong y học
Prasugrel được dùng phối hợp với aspirin liều thấp để ngăn ngừa huyết khối ở những người bệnh mắc hội chứng mạch vành cấp tính, bao gồm đau thắt ngực không ổn định, nhồi máu cơ tim không ST chênh lên (NSTEMI), nhồi máu cơ tim ST chênh lên (STEMI) và những người được lên kế hoạch điều trị bằng PCI.
Nguy cơ chảy máu liên quan đến prasugrel cao hơn so với clopidogrel nhưng nó đã chứng minh được tính ưu việt trong việc giảm tiêu chí gộp là tử vong, nhồi máu cơ tim tái phát và đột quỵ. Tuy nhiên, prasugrel không làm thay đổi nguy cơ tử vong khi dùng cho những người bị STEMI hoặc NSTEMI.
Dược động học
Hấp thu
Sau khi uống, khoảng 79% hoặc cao hơn liều dùng được hấp thu và sau đó được chuyển hóa nhanh chóng. Nồng độ đỉnh trong huyết tương (Cmax) đạt được trong khoảng 30 phút sau khi uống. Khi uống cùng với bữa ăn nhiều chất béo, nhiều calo không ảnh hưởng đến AUC của chất chuyển hóa có hoạt tính ở những người khỏe mạnh, nhưng Cmax giảm ~49% và Tmax tăng lên 0,5 – 1,5 giờ. Prasugrel có thể được dùng cùng hoặc không cùng thức ăn.
Phân bố
Khoảng 98% chất chuyển hóa có hoạt tính được liên kết với albumin huyết thanh người trong dung dịch đệm 4%. Các chất chuyển hóa chính không có hoạt tính cũng liên kết cao với protein huyết tương người. Thể tích phân bố của prasugrel là 44-68L.
Sự phân bố mô của hoạt tính phóng xạ liên quan đến prasugrel đã được nghiên cứu ở chuột sau khi uống một lần và lặp lại. Phóng xạ được phân phối rộng rãi và nhanh chóng khắp cơ thể. Nồng độ phóng xạ cao nhất ở hầu hết các mô tham gia vào quá trình hấp thụ và loại bỏ hợp chất và các chất chuyển hóa của nó, bao gồm dạ dày, ruột, gan, thận và bàng quang. Prasugrel phân bố vào tủy xương của chuột cống với tỷ lệ giữa mô và huyết tương nhỏ hơn 0,5.
Sau khi dùng liều lặp lại hàng ngày, sự tích lũy phù hợp với thời gian bán thải của prasugrel được quan sát thấy ở hầu hết các cơ quan.
Chuyển hóa
Prasugrel không được phát hiện trong huyết tương sau khi uống. Nó nhanh chóng bị thủy phân trong ruột thành thiolactone bởi carboxylesterase của con người (hCE)2. Chất trung gian này được chuyển hóa tiếp thành chất chuyển hóa có hoạt tính của nó là R-138727, trong một bước duy nhất bởi các enzym cytochrom P450 trong gan (chủ yếu là CYP3A4 và CYP2B6 và thành một mức độ thấp hơn bởi CYP2C9 và CYP2C19).
Chất chuyển hóa có hoạt tính được tiếp tục chuyển hóa bằng cách S-methyl hóa hoặc liên hợp cysteine thành hai chất chuyển hóa không có hoạt tính. Không giống như clopidogrel, sự biến đổi prasugrel thành chất chuyển hóa có hoạt tính dường như không bị ảnh hưởng bởi tính đa hình của cytochrom P450.
Thải trừ
Khoảng 68% liều uống prasugrel được bài tiết qua nước tiểu và 27% qua phân dưới dạng các chất chuyển hóa không có hoạt tính. Chất chuyển hóa có hoạt tính dự kiến sẽ không bị loại bỏ bằng thẩm tách.
Chất chuyển hóa có hoạt tính có thời gian bán thải khoảng 7,4 giờ (trong khoảng 2-15 giờ). Độ thanh thải biểu kiến khoảng 112 – 166 L/giờ.
Độc tính ở người
Thienopyridin, bao gồm cả prasugrel, làm tăng nguy cơ chảy máu. Với chế độ dùng thuốc được sử dụng trong TRITONTIMI 38, TIMI (Huyết khối trong nhồi máu cơ tim) nặng (xuất huyết rõ ràng trên lâm sàng liên quan đến giảm huyết sắc tố ≥5 g/dL, hoặc xuất huyết nội sọ) và các biến cố chảy máu nhẹ TIMI (chảy máu quá mức liên quan đến giảm huyết sắc tố ≥3 g/dL nhưng <5 g/dL) phổ biến hơn ở nhóm prasugrel so với nhóm clopidogrel.
Tính an toàn
Thời kỳ mang thai
Không có nghiên cứu đầy đủ và được kiểm soát tốt về việc sử dụng prasugrel hiệu quả ở phụ nữ mang thai.
Các nghiên cứu về độc tính sinh sản và phát triển ở chuột và thỏ với liều gấp 30 lần mức phơi nhiễm điều trị được khuyến nghị ở người (dựa trên sự phơi nhiễm huyết tương với chất chuyển hóa chính lưu hành ở người) cho thấy không có bằng chứng gây hại cho bào thai; tuy nhiên, các nghiên cứu trên động vật không phải lúc nào cũng dự đoán được phản ứng của con người. Do đó, prasugrel chỉ nên được sử dụng trong thời kỳ mang thai nếu lợi ích tiềm năng cho người mẹ lớn hơn nguy cơ tiềm ẩn đối với thai nhi.
Trong các nghiên cứu về độc tính đối với sự phát triển của phôi thai, chuột cống và thỏ mang thai dùng prasugrel ở liều uống gây độc cho chuột mẹ tương đương hơn 40 lần so với liều dùng ở người. Người ta quan sát thấy trọng lượng cơ thể của thai nhi giảm nhẹ; nhưng, không có dị tật cấu trúc ở cả hai loài.
Trong các nghiên cứu trên chuột trước khi sinh và sau khi sinh, việc điều trị bằng prasugrel ở chuột mẹ không ảnh hưởng đến sự phát triển hành vi hoặc sinh sản của chuột con ở liều cao hơn 150 lần so với liều dùng ở người.
Thời kỳ cho con bú
Không biết liệu prasugrel có bài tiết qua sữa mẹ hay không; tuy nhiên, các chất chuyển hóa của prasugrel được tìm thấy trong sữa chuột. Do nhiều thuốc được bài tiết qua sữa mẹ, chỉ nên sử dụng prasugrel trong thời kỳ cho con bú nếu lợi ích cho người mẹ lớn hơn nguy cơ có thể xảy ra cho trẻ bú mẹ.
Trẻ em
Tính an toàn và hiệu quả của prasugrel ở bệnh nhi chưa được thiết lập,
Người cao tuổi
Do nguy cơ chảy máu (kể cả chảy máu gây tử vong) và hiệu quả không chắc chắn ở bệnh nhân trên 75 tuổi, việc sử dụng prasugrel thường không được khuyến cáo ở những bệnh nhân này, ngoại trừ những trường hợp có nguy cơ cao (bệnh nhân đái tháo đường hoặc tiền sử nhồi máu cơ tim) thì có thể cân nhắc lợi việc sử dụng thuốc.
Suy thận
Không cần điều chỉnh liều cho bệnh nhân suy thận. Mặc dù kinh nghiệm điều trị bệnh nhân mắc bệnh thận giai đoạn cuối còn hạn chế, nhưng những bệnh nhân này thường có nguy cơ chảy máu cao hơn.
Suy gan
Không cần điều chỉnh liều ở bệnh nhân suy gan nhẹ đến trung bình. Mặc dù dược động học và dược lực học của prasugrel ở những bệnh nhân mắc bệnh gan nặng chưa được nghiên cứu, nhưng những bệnh nhân này thường có nguy cơ chảy máu cao hơn.
Ung thư
Không quan sát thấy khối u liên quan đến hợp chất nào trong một nghiên cứu trên chuột trong 2 năm với prasugrel ở liều uống lên đến 100 mg/kg/ngày (>100 lần so với nồng độ điều trị khuyến cáo ở người (dựa trên phơi nhiễm huyết tương với chất chuyển hóa chính lưu hành ở người). Tuy nhiên, có sự gia tăng tỷ lệ mắc các khối u (u tuyến tế bào gan) ở chuột tiếp xúc với liều cao trong 2 năm (>250 lần so với tiếp xúc với chất chuyển hóa ở người).
Đột biến
Prasugrel không gây độc tính di truyền trong hai thử nghiệm in vitro (thử nghiệm đột biến gen vi khuẩn Ames, thử nghiệm tạo clastogen ở nguyên bào sợi của chuột đồng Trung Quốc) và trong một thử nghiệm in vivo (thử nghiệm vi nhân bằng đường trong phúc mạc ở chuột nhắt).
Tương tác với thuốc khác
Prasugrel được chuyển hóa bởi hệ thống enzyme microsome cytochrom P-450 (CYP), chủ yếu bởi isoenzyme 3A4 và 2B6, và ở mức độ thấp hơn bởi isoenzyme 2C9 và 2C19. Các nghiên cứu in vitro chỉ ra rằng prasugrel không có khả năng ức chế các isoenzym của CYP 1A2, 2C9, 2C19, 2D6 và 3A hoặc cảm ứng các isoenzym của CYP 1A2 hoặc 3A. Đồng thời, prasugrel là chất ức chế yếu CYP2B6.
Các tương tác thuốc quan trọng trên lâm sàng qua trung gian các isoenzym CYP được coi là khó xảy ra với prasugrel. Các nghiên cứu đã chỉ ra rằng nếu một trong các isoenzym của CYP tham gia vào quá trình chuyển hóa của prasugrel bị ức chế, thì các isoenzym khác vẫn có khả năng tạo thành chất chuyển hóa có hoạt tính.
Các chất ức chế CYP3A4 như verapamil, diltiazem, indinavir, ciprofloxacin, clarithromycin và nước ép bưởi được cho là không có ảnh hưởng đáng kể đến dược động học của chất chuyển hóa có hoạt tính của prasugrel. Ở những người khỏe mạnh dùng prasugrel, dùng đồng thời với ketoconazole (một chất ức chế mạnh CYP3A4) làm giảm nồng độ tối đa của chất chuyển hóa có hoạt tính của prasugrel khoảng 34-46%, nhưng không làm thay đổi nồng độ toàn thân hoặc mức độ ức chế kết tập tiểu cầu.
Tương tự, chất cảm ứng CYP3A4 (ví dụ, rifampin, carbamazepine) không được cho là sẽ làm thay đổi đáng kể đáp ứng dược động học hoặc dược lực học với prasugrel; dùng đồng thời với rifampin, một chất gây cảm ứng mạnh CYP3A4 và CYP2B6, không ảnh hưởng đến sự hình thành chất chuyển hóa có hoạt tính của prasugrel cũng như khả năng ức chế kết tập tiểu cầu của nó.
Sử dụng đồng thời prasugrel và các thuốc được chuyển hóa chủ yếu bởi CYP2B6 (ví dụ: halothane, cyclophosphamide, propofol, nevirapine) có thể làm tăng nồng độ trong huyết tương và mức độ tiếp xúc của thuốc dùng đồng thời; tuy nhiên, các tương tác này không quan trọng về mặt lâm sàng do prasugrel ức chế yếu đối với CYP2B6.
Sử dụng đồng thời prasugrel và warfarin hoặc NSAID (được sử dụng lâu dài) có thể làm tăng nguy cơ chảy máu.
Prasugrel có thể được sử dụng đồng thời với aspirin (75 mg đến 325 mg mỗi ngày), heparin, thuốc ức chế GPIIb/IIIa, statin, digoxin và thuốc làm tăng pH dạ dày, bao gồm thuốc ức chế bơm proton và thuốc chẹn H2.
Lưu ý khi sử dụng Prasugrel
Prasugrel được chống chỉ định ở những bệnh nhân đang chảy máu bệnh lý như loét dạ dày tá tràng hoặc xuất huyết nội sọ; những bệnh nhân có tiền sử cơn thiếu máu não thoáng qua (TIA) hoặc đột quỵ.
Bệnh nhân bị đột quỵ hoặc TIA khi đang dùng prasugrel nói chung nên ngừng điều trị.
Thienopyridin ức chế kết tập tiểu cầu trong suốt thời gian tồn tại của tiểu cầu (710 ngày), do đó, việc giữ lại một liều sẽ không hữu ích trong việc kiểm soát biến cố chảy máu hoặc nguy cơ chảy máu liên quan đến thủ thuật xâm lấn.
Vì thời gian bán hủy của chất chuyển hóa có hoạt tính của prasugrel ngắn so với thời gian tồn tại của tiểu cầu, nên có thể khôi phục lại quá trình cầm máu bằng cách sử dụng tiểu cầu ngoại sinh; tuy nhiên, truyền tiểu cầu trong vòng 6 giờ sau liều tấn công hoặc 4 giờ sau liều duy trì có thể kém hiệu quả hơn.
Nguy cơ chảy máu tăng lên ở những bệnh nhân dùng prasugrel trải qua phẫu thuật ghép bắc cầu động mạch vành (CABG). Nếu có thể, nên ngừng prasugrel ít nhất 7 ngày trước CABG.
Ở những bệnh nhân được điều trị bằng PCI và đặt stent, việc ngừng sớm bất kỳ loại thuốc chống kết tập tiểu cầu nào, kể cả thienopyridin, sẽ làm tăng nguy cơ huyết khối trong stent, nhồi máu cơ tim và tử vong. Những bệnh nhân cần ngừng thienopyridine sớm sẽ tăng nguy cơ mắc các biến cố tim mạch. Cần tránh tình trạng mất hiệu lực trong điều trị và nếu thienopyridin phải tạm thời ngừng sử dụng vì các tác dụng phụ, thì chúng nên được bắt đầu lại càng sớm càng tốt.
Ban xuất huyết giảm tiểu cầu huyết khối (TTP) đã được báo cáo khi sử dụng prasugrel. TTP có thể xảy ra sau một thời gian ngắn tiếp xúc (<2 tuần). TTP là một tình trạng nghiêm trọng có thể gây tử vong và cần điều trị khẩn cấp, được đặc trưng bởi giảm tiểu cầu, thiếu máu tán huyết vi mạch (tế bào phiến [tế bào hồng cầu mảnh] nhìn thấy trên phết máu ngoại vi), phát hiện thần kinh, rối loạn chức năng thận và sốt.
Những người có trọng lượng cơ thể <60 kg tăng nguy cơ chảy máu và tăng phơi nhiễm với chất chuyển hóa có hoạt tính của prasugrel. Do đó, cần cân nhắc giảm liều duy trì xuống 5 mg ở bệnh nhân <60 kg. Tuy nhiên, hiệu quả và độ an toàn của liều 5 mg chưa được nghiên cứu tiền cứu.
Một vài nghiên cứu của Prasugrel trong Y học
Thuốc kháng kết tập tiểu cầu để thuyên tắc phình mạch nội sọ được hỗ trợ bằng cuộn dây stent
Đặt vấn đề: Việc sử dụng thuốc chống kết tập tiểu cầu để ngăn ngừa huyết khối trong điều trị phình động mạch não bằng stent đã được nhấn mạnh rộng rãi.
Mục tiêu: So sánh prasugrel liều thấp với clopidogrel trong thuyên tắc phình mạch nội sọ bằng ống thông có hỗ trợ đặt stent.
Phương pháp: Đây là một đánh giá hồi cứu 311 chứng phình động mạch từ 297 bệnh nhân đã trải qua thuyên tắc nội mạch bằng cuộn dây có hỗ trợ đặt stent cho chứng phình động mạch nội sọ chưa vỡ trong khoảng thời gian từ tháng 11 năm 2014 đến tháng 3 năm 2017.
Các biến cố bất lợi do huyết khối và xuất huyết được so sánh giữa 207 bệnh nhân dùng prasugrel liều thấp (PSG) nhóm) và 90 bệnh nhân dùng clopidogrel (nhóm CPG).
Kết quả: Giá trị đơn vị phản ứng P2Y12 (PRU) thấp hơn đáng kể trong nhóm PSG (nhóm PSG so với nhóm CPG, 132,3 ± 76,9 so với 238,1 ± 69,1; P <0,001); tỷ lệ ức chế cũng cao hơn về mặt thống kê ở nhóm PSG (54,0 ± 26,0% so với 20,8 ± 18,6%; P < 0,001).
Các biến cố huyết khối tắc mạch xảy ra ít thường xuyên hơn ở nhóm PSG so với nhóm CPG (0,9% so với 6,4%; P = 0,01), trong khi không có sự khác biệt đáng kể về tỷ lệ biến chứng xuất huyết (0,5% so với 2,2%; P = 0,22 ).
Trong phân tích đa biến, clopidogrel với vai trò là thuốc kháng tiểu cầu là yếu tố nguy cơ đáng kể duy nhất đối với thuyên tắc huyết khối ở loạt bệnh nhân này trải qua thuyên tắc ống thông có hỗ trợ đặt stent.
Kết luận: Sử dụng PSG liều thấp như một thuốc kháng tiểu cầu là nhanh chóng, hiệu quả và an toàn để thuyên tắc bằng cuộn dây hỗ trợ đặt stent của phình động mạch nội sọ chưa vỡ. Tiền mê Prasugrel làm giảm đáng kể tần suất các biến cố huyết khối tắc mạch mà không làm tăng nguy cơ xuất huyết.
Tài liệu tham khảo
- Choi HH, Lee JJ, Cho YD, Han MH, Cho WS, Kim JE, An SJ, Mun JH, Yoo DH, Kang HS. Antiplatelet Premedication for Stent-Assisted Coil Embolization of Intracranial Aneurysms: Low-Dose Prasugrel vs Clopidogrel. Neurosurgery. 2018 Nov 1;83(5):981-988. doi: 10.1093/neuros/nyx591. PMID: 29301051.
- Drugbank, Prasugrel, truy cập ngày 14 tháng 4 năm 2023.
- Pubchem, Prasugrel, truy cập ngày 14 tháng 4 năm 2023.
- Bộ Y Tế (2012), Dược thư quốc gia Việt Nam, Nhà xuất bản Y học, Hà Nội
Xuất xứ: Đức
Xuất xứ: Việt Nam

Xuất xứ: Mỹ