


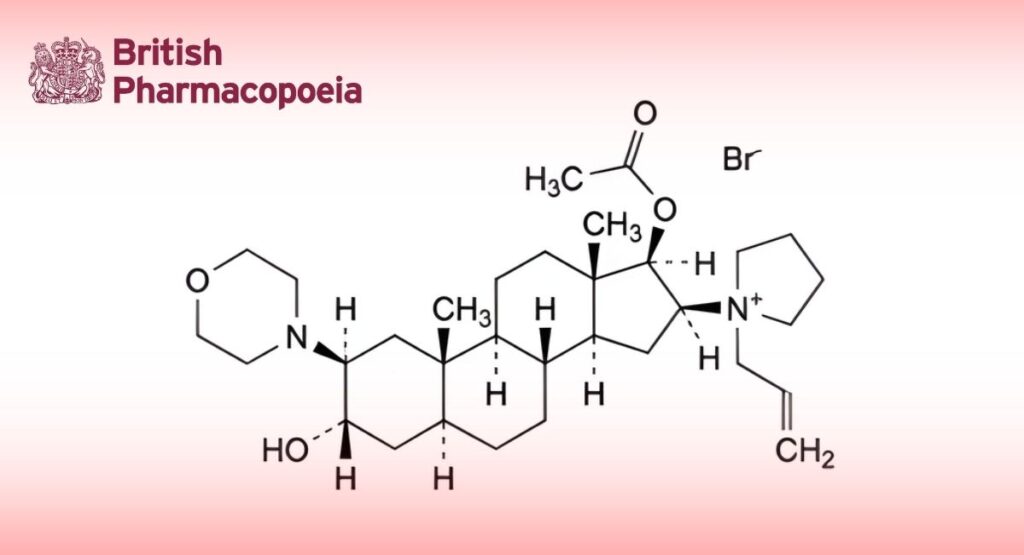
(Ph. Eur. monograph 1764)
C32H53BrN2O4 610 119302-91-9
Action and use
Non-depolarizing neuromuscular blocker.
DEFINITION
1-[17β-Acetoxy-3α-hydroxy-2β-(morpholin-4-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium bromide.
Content
99.0 per cent to 101.0 per cent (anhydrous substance).
CHARACTERS
Appearance
Almost white or pale yellow, slightly hygroscopic powder.
Solubility
Freely soluble in water, very soluble in methylene chloride, freely soluble in anhydrous ethanol.
IDENTIFICATION
A. Infrared absorption spectrophotometry (2.2.24).
Comparison: rocuronium bromide CRS.
B. Solution S (see Tests) gives reaction (a) of bromides (2.3.1).
TESTS
Solution S
Dissolve 0.10 g in carbon dioxide-free water R and dilute to 10 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and not more intensely coloured than reference solution BY5 (2.2.2, Method II).
Specific optical rotation (2.2.7)
+ 28.5 to + 32.0 (anhydrous substance).
Dissolve 0.250 g in a 5.15 g/L solution of hydrochloric acid R and dilute to 25.0 mL with the same solution.
pH (2.2.3)
8.9 to 9.5 for solution S.
Related substances
Liquid chromatography (2.2.29).
Solvent mixture water R, acetonitrile R1 (10:90 V/V).
Test solution: Dissolve 0.100 g of the substance to be examined in the solvent mixture and dilute to 10.0 mL with the solvent mixture.
Reference solution (a): Dilute 1.0 mL of the test solution to 100.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b): Dissolve 5 mg of rocuronium for peak identification CRS (containing impurities A, B, C, F, G and H) in the solvent mixture and dilute to 5.0 mL with the solvent mixture.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: silica gel for chromatography R (5 μm);
— temperature: 30 °C.
Mobile phase: Mix 10 volumes of a 4.53 g/L solution of tetramethylammonium hydroxide R adjusted to pH 7.4 with phosphoric acid R and 90 volumes of acetonitrile R1.
Flow rate: 2.0 mL/min.
Detection: Spectrophotometer at 210 nm.
Injection: 5 μL.
Run time: 2.5 times the retention time of rocuronium.
Identification of impurities: Use the chromatogram supplied with rocuronium for peak identification CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities A, B, C, F, G and H.
Relative retention: With reference to rocuronium (retention time = about 9 min): impurity A = about 0.2; impurity G = about 0.4; impurity F = about 0.75; impurity B = about 0.80; impurity H = about 0.95; impurity C = about 1.2.
System suitability: Reference solution (b):
— peak-to-valley ratio: minimum 3.0, where Hp = height above the baseline of the peak due to impurity H and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to rocuronium.
Limits:
— correction factors: for the calculation of content, multiply the peak areas of the following impurities by the corresponding correction factor: impurity A = 0.5; impurity F = 1.3; impurity G = 0.4; impurity H = 0.4;
— impurities A, B, C: for each impurity, not more than twice the area of the principal peak in the chromatogram obtained with reference solution (a) (0.2 per cent);
— impurities F, G, H: for each impurity, not more than 1.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.15 per cent);
— unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (a) (0.10 per cent);
— total: not more than 10 times the area of the principal peak in the chromatogram obtained with reference solution (a) (1.0 per cent);
— disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.05 per cent); disregard any peak eluting before impurity A.
Chlorides
Liquid chromatography (2.2.29).
Test solution: Dissolve 20.0 mg of the substance to be examined in water R and dilute to 20.0 mL with the same solvent.
Reference solution (a): Dissolve 0.644 g of sodium bromide R and 0.824 g of sodium chloride R in water R and dilute to 1000.0 mL with the same solvent. Dilute 1.0 mL of the solution to 50.0 mL with water R.
Reference solution (b): Dissolve 0.824 g of sodium chloride R in water R and dilute to 1000.0 mL with the same solvent. Dilute 5.0 mL of the solution to 50.0 mL with water R. Dilute 2.0 mL of this solution to 50.0 mL with water R.
Blank solution water R.
Precolumn:
— size: l = 0.05 m, Ø = 4.0 mm;
— stationary phase: anion-exchange resin R (13 μm).
Column:
— size: l = 0.25 m, Ø = 4.0 mm;
— stationary phase: anion-exchange resin R (13 μm).
Mobile phase: A solution containing 0.063 g/L of sodium hydrogen carbonate R and 0.212 g/L of anhydrous sodium carbonate R.
Flow rate: 2.0 mL/min.
Detection: Conductivity detector set at 100 μS/V and maintained at 30 °C.
Use a self-regenerating anion suppressor.
Injection: 25 μL.
Retention times: Chloride = about 1.7 min; bromide = about 2.8 min.
System suitability: Reference solution (a):
— resolution: minimum 2.5 between the peaks due to chloride and bromide.
Limit:
— chlorides: not more than 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.1 per cent).
2-Propanol (2.4.24, System A)
Maximum 1.0 per cent.
Water (2.5.12)
Maximum 4.5 per cent, determined on 0.400 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.400 g in 40 mL of glacial acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 60.97 mg of C32H53BrN2O4.
STORAGE
In an airtight container, protected from light, at a temperature below -15 °C.
IMPURITIES
Specified impurities A, B, C, F, G, H.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) D, E.
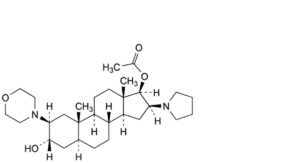
A. 3α-hydroxy-2β-(morpholin-4-yl)-16β-(pyrrolidin-1-yl)-5α-androstan-17β-yl acetate,
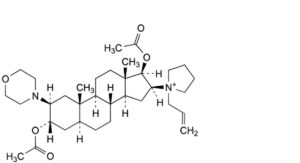
B. 1-[3α,17β-diacetoxy-2β-(morpholin-4-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium,
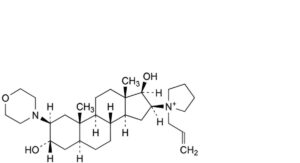
C. 1-[3α,17β-dihydroxy-2β-(morpholin-4-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium,
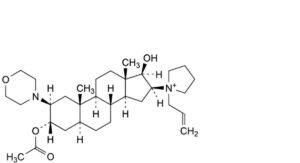
D. 1-[3α-acetoxy-17β-hydroxy-2β-(morpholin-4-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium,
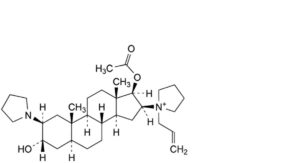
E. 1-[17β-acetoxy-3α-hydroxy-2β-(pyrrolidin-1-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium,
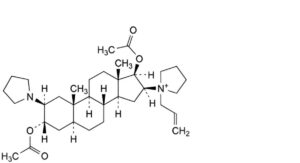
F. 1-[3α,17β-acetoxy-2β-(pyrrolidin-1-yl)-5α-androstan-16β-yl]-1-(prop-2-enyl)pyrrolidinium,
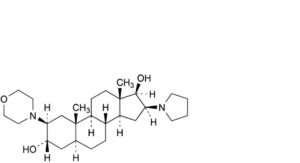
G. 2β-(morpholin-4-yl)-16β-(pyrrolidin-1-yl)-5α-androstane-3α,17β-diol,
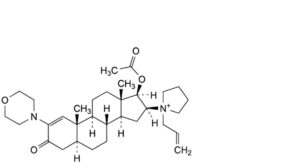
H. 1-[17β-acetoxy-2-(morpholin-4-yl)-3-oxo-5α-androst-1-en-16β-yl]-1-(prop-2-enyl)pyrrolidinium.


