


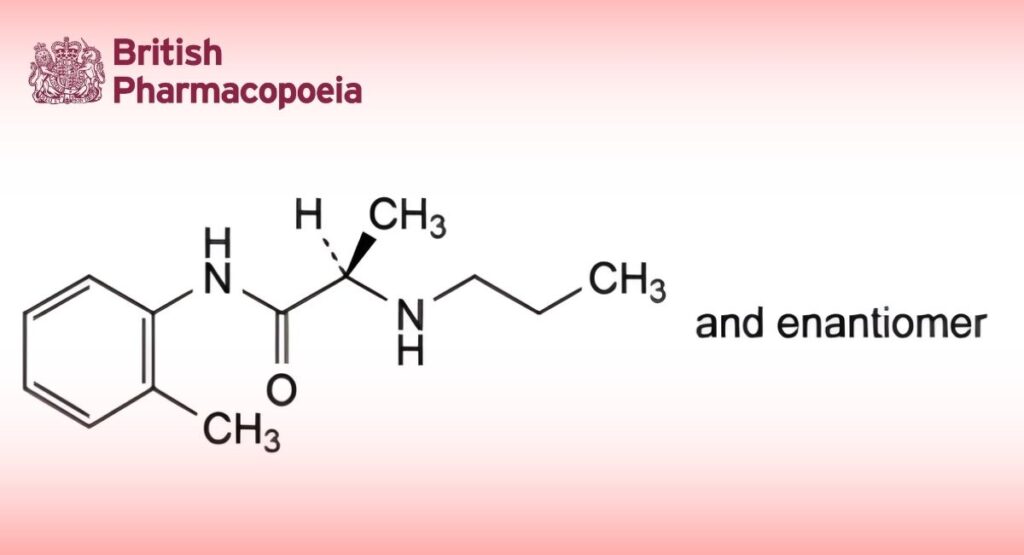
(Ph. Eur. monograph 1362)
C13H20N2O 220.3 721-50-6
Action and use
Local anaesthetic.
DEFINITION
(2RS)-N-(2-Methylphenyl)-2-(propylamino)propanamide.
Content
99.0 per cent to 101.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Slightly soluble in water, very soluble in acetone and in ethanol (96 per cent).
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Preparation: Prepare a film between 2 plates of sodium chloride R by heating at 40-45 °C until the substance has melted.
Comparison: prilocaine CRS.
TESTS
Appearance of solution
The solution is clear (2.2.1) and colourless (2.2.2, Method II).
Dissolve 2.50 g in 15 mL of dilute hydrochloric acid R and dilute to 50.0 mL with water R.
Related substances
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Test solution: Dissolve 25 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a): Dissolve 2.5 mg of the substance to be examined and 3 mg of prilocaine impurity E CRS in the mobile phase and dilute to 100.0 mL with the mobile phase. Dilute 1.0 mL of the solution to 10.0 mL with the mobile phase.
Reference solution (b): Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (c): Dissolve 33.5 mg of prilocaine impurity B CRS in the mobile phase and dilute to 100.0 mL with the mobile phase. Dilute 1.0 mL of the solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (d): Dissolve 15 mg of prilocaine for peak identification CRS (containing impurity G) in the mobile phase and dilute to 5.0 mL with the mobile phase.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: end-capped extra-dense bonded octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase: Mix 26 volumes of acetonitrile for chromatography R and 74 volumes of a solution prepared as follows: dissolve 0.180 g of sodium dihydrogen phosphate monohydrate R and 2.89 g of disodium hydrogen phosphate dihydrate R in 1000 mL of water for chromatography R.
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 240 nm.
Injection: 20 μL.
Run time: Twice the retention time of prilocaine.
Identification of impurities: Use the chromatogram obtained with reference solution (c) to identify the peak due to impurity B; use the chromatogram obtained with reference solution (a) to identify the peak due to impurity E; use the chromatogram obtained with reference solution (d) to identify the peak due to impurity G.
Relative retention: With reference to prilocaine (retention time = about 25 min): impurity B = about 0.3; impurity G = about 0.8; impurity E = about 1.2.
System suitability: Reference solution (a):
— resolution: minimum 3.0 between the peaks due to prilocaine and impurity E.
Limits:
— impurity B: not more than the area of the corresponding peak in the chromatogram obtained with reference solution (c) (100 ppm);
— impurity G: not more than 1.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.15 per cent);
— unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (0.10 per cent);
— total: not more than twice the area of the principal peak in the chromatogram obtained with reference solution (b) (0.2 per cent);
— disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.05 per cent).
Water (2.5.12)
Maximum 0.5 per cent, determined on 1.000 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.180 g in 50 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 22.03 mg of C13H20N2O.
IMPURITIES
Specified impurities B, G.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) A, C, D, E, F.
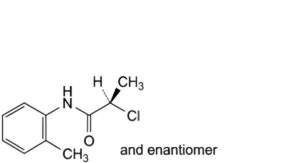
A. (2RS)-2-chloro-N-(2-methylphenyl)propanamide,
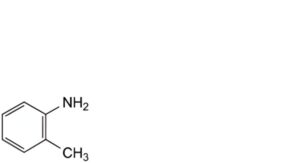
B. 2-methylbenzenamine (o-toluidine),
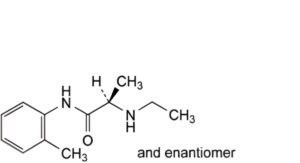
C. (2RS)-2-(ethylamino)-N-(2-methylphenyl)propanamide,
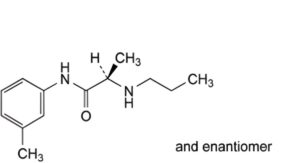
D. (2RS)-N-(3-methylphenyl)-2-(propylamino)propanamide,
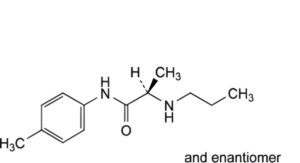
E. (2RS)-N-(4-methylphenyl)-2-(propylamino)propanamide,
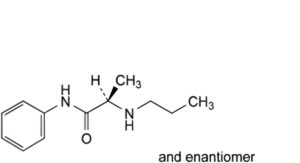
F. (2RS)-N-phenyl-2-(propylamino)propanamide,
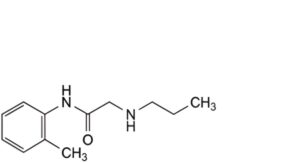
G. N-(2-methylphenyl)-2-(propylamino)acetamide.


