


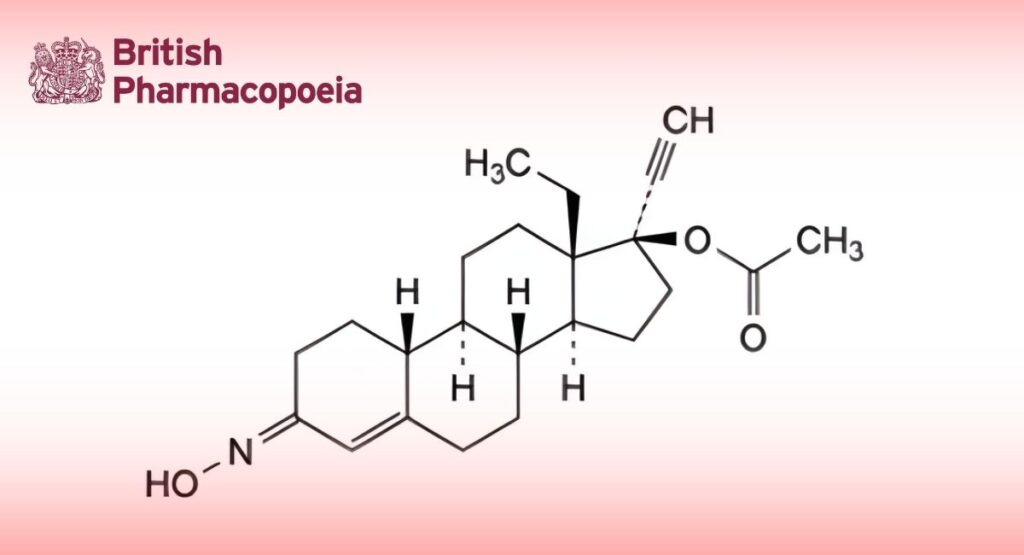
(Ph. Eur. monograph 1732)
C23H31NO3 369.5 35189-28-7
Action and use
Progestogen.
DEFINITION
(3EZ)-13β-Ethyl-3-(hydroxyimino)-18,19-dinor-17α-pregn-4-en-20-yn-17-yl acetate.
Content
98.5 per cent to 101.5 per cent (dried substance).
CHARACTERS
Appearance
White or almost white powder.
Solubility
Practically insoluble in water, freely soluble in methylene chloride, soluble in acetone.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparison: norgestimate CRS.
TESTS
Specific optical rotation (2.2.7)
+ 42.0 to + 50.0 (dried substance).
Dissolve 0.200 g in methylene chloride R and dilute to 20.0 mL with the same solvent.
Related substances
Liquid chromatography (2.2.29).
Solvent mixture water R, methanol R (1:4 V/V).
Test solution: Dissolve 25.0 mg of the substance to be examined in the solvent mixture and dilute to 50.0 mL with the solvent mixture.
Reference solution (a): Dilute 2.0 mL of the test solution to 100.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 20.0 mL with the solvent mixture.
Reference solution (b): Dissolve 2 mg of norgestimate for system suitability CRS (containing impurity A) in 4 mL of the solvent mixture.
Column:
— size: l = 0.10 m, Ø = 4.6 mm;
— stationary phase: spherical end-capped octadecylsilyl silica gel for chromatography R (5 μm);
— temperature: 40 °C.
Mobile phase: acetonitrile R, tetrahydrofuran for chromatography R, water R (18:22:60 V/V/V).
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 244 nm.
Injection: 25 μL.
Run time: Twice the retention time of the (E)-isomer of norgestimate.
Identification of impurities: Use the chromatogram supplied with norgestimate for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peak due to impurity A.
Relative retention: With reference to the (E)-isomer of norgestimate (retention time = about 14 min): impurity A = about 0.7; (Z)-isomer of norgestimate = about 0.9.
System suitability: Reference solution (b):
— resolution: minimum 1.5 between the peaks due to the (E)- and (Z)-isomers of norgestimate.
Limits:
— correction factor: for the calculation of content, multiply the peak area of the (Z)-isomer of norgestimate by 1.33;
— impurity A: not more than twice the sum of the areas of the peaks due to the (E)- and (Z)-isomers of norgestimate in the chromatogram obtained with reference solution (a) (0.2 per cent);
— unspecified impurities: for each impurity, not more than the sum of the areas of the peaks due to the (E)- and (Z)-isomers of norgestimate in the chromatogram obtained with reference solution (a) (0.10 per
cent);
— total: not more than 3 times the sum of the areas of the peaks due to the (E)- and (Z)-isomers of norgestimate in the chromatogram obtained with reference solution (a) (0.3 per cent);
— disregard limit: 0.5 times the sum of the areas of the peaks due to the (E)- and (Z)-isomers of norgestimate in the chromatogram obtained with reference solution (a) (0.05 per cent).
Ratio of (E)- to (Z)-isomers
Liquid chromatography (2.2.29) as described in the test for related substances with the following modification.
Injection: Test solution.
Calculate the (E)- to (Z)-isomer ratio by dividing the area of the peak due to the (E)-isomer by 1.33 times the area of the peak due to the (Z)-isomer. The ratio is 1.27 to 1.78.
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on 0.500 g by drying in an oven at 105 °C for 3 h.
ASSAY
Dissolve 0.300 g in 40 mL of tetrahydrofuran R. Add 10 mL of a 100 g/L solution of silver nitrate R and titrate with 0.1 M sodium hydroxide, determining the end-point potentiometrically (2.2.20). Rinse the electrode with
acetone R after each titration.
If necessary, after several titrations re-equilibrate the electrode in water R for 15 min to obtain sharper titration curves.
1 mL of 0.1 M sodium hydroxide is equivalent to 36.95 mg of C23H31NO3.
IMPURITIES
Specified impurities A.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for
other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10.
Control of impurities in substances for pharmaceutical use) B, C, D.
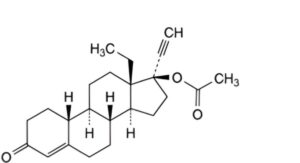
A. 13β-ethyl-3-oxo-18,19-dinor-17α-pregn-4-en-20-yn-17-yl acetate (levonorgestrel acetate),
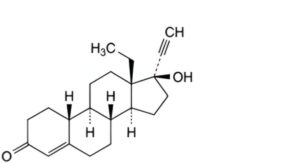
B. 13β-ethyl-17β-hydroxy-18,19-dinor-17α-pregn-4-en-20-yn-3-one (levonorgestrel),
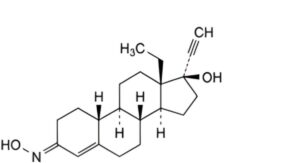
C. (3E)-13β-ethyl-3-(hydroxyimino)-18,19-dinor-17α-pregn-4-en-20-yn-17-ol ((E)-norelgestromin),
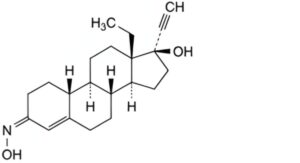
D. (3Z)-13β-ethyl-3-(hydroxyimino)-18,19-dinor-17α-pregn-4-en-20-yn-17-ol ((Z)-norelgestromin).


