


Edition: BP 2025 (Ph. Eur. 11.6 update)
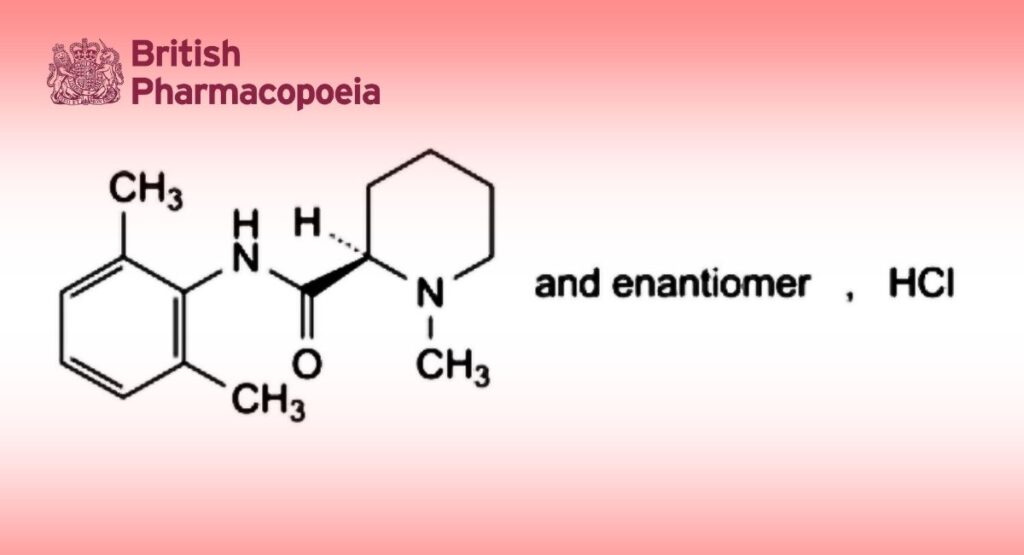
C15H23ClN2O 282.8 1722-62-9
Action and use
Local anaesthetic.
DEFINITION
(2RS)-N-(2,6-Dimethylphenyl)-1-methylpiperidine-2-carboxamide hydrochloride.
Content
98.5 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Freely soluble in water and in ethanol (96 per cent), very slightly soluble in methylene chloride.
mp
About 260 °C, with decomposition.
It shows polymorphism (5.9).
IDENTIFICATION
First identification: A, C.
Second identification: B, C.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison mepivacaine hydrochloride CRS.
If the spectra obtained in the solid state show differences, dissolve the substance to be examined and the reference substance separately in methanol R, evaporate to dryness and dry in an oven at 80 °C for 45 min. Record new spectra using the residues.
B. Thin-layer chromatography (2.2.27).
Test solution Dissolve 20 mg of the substance to be examined in ethanol (96 per cent) R and dilute to 5 mL with the same solvent.
Reference solution (a) Dissolve 20 mg of mepivacaine hydrochloride CRS in ethanol (96 per cent) R and dilute to 5 mL with the same solvent.
Reference solution (b) Dissolve 20 mg of mepivacaine hydrochloride CRS and 20 mg of lidocaine hydrochloride CRS in ethanol (96 per cent) R and dilute to 5 mL with the same solvent.
Plate TLC silica gel F254 plate R.
Mobile phase concentrated ammonia R, methanol R, 1,1-dimethylethyl methyl ether R (1:5:100 V/V/V).
Application 10 μL.
Development Over 2/3 of the plate.
Drying In air.
Detection Examine in ultraviolet light at 254 nm.
System suitability Reference solution (b):
— the chromatogram shows 2 clearly separated principal spots.
Results The principal spot in the chromatogram obtained with the test solution is similar in position and size to the principal spot in the chromatogram obtained with reference solution (a).
C. It gives reaction (a) of chlorides (2.3.1).
TESTS
Solution S
Dissolve 1.500 g in carbon dioxide-free water R and dilute to 30.0 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and not more intensely coloured than reference solution B7
(2.2.2, Method II).
pH (2.2.3)
4.0 to 5.0.
Dilute 2 mL of solution S to 5 mL with carbon dioxide-free water R.
Optical rotation (2.2.7)
-0.10° to + 0.10°, determined on solution S.
Related substances
Liquid chromatography (2.2.29).
Test solution Dissolve 20.0 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a) Dissolve 20.0 mg of the substance to be examined and 30.0 mg of mepivacaine impurity B CRS in the mobile phase and dilute to 100.0 mL with the mobile phase. Dilute 1.0 mL of the solution to 100.0 mL with the mobile phase.
Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Column:
— size: l = 0.125 m, Ø = 4.6 mm;
— stationary phase: base-deactivated end-capped octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase Mix 35 volumes of acetonitrile R1 and 65 volumes of a 2.25 g/L solution of phosphoric acid R, previously adjusted to pH 7.6 with strong sodium hydroxide solution R.
Flow rate 1 mL/min.
Detection Spectrophotometer at 220 nm.
Injection 20 μL.
Run time 3 times the retention time of mepivacaine.
Identification of impurities Use the chromatogram obtained with reference solution (a) to identify the peak due to impurity B.
Relative retention With reference to mepivacaine (retention time = about 7 min): impurity B = about 0.5.
System suitability Reference solution (a):
— resolution: minimum 2.5 between the peaks due to impurity B and mepivacaine.
Limits:
— unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (0.10 per cent);
— total: not more than 5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.5 per cent);
— disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.05 per cent).
Impurity A
Head-space gas chromatography (2.2.28).
Test solution Introduce 60.0 mg of the substance to be examined into a 20 mL vial. Add 2.0 mL of a 103.0 g/L solution of hydrochloric acid R and 1.0 mL of a 126.0 g/L solution of sodium hydroxide R, and close the vial immediately.
Reference solution Dissolve 6.0 mg of bupivacaine impurity F CRS (impurity A) in a 103.0 g/L solution of hydrochloric acid R and dilute to 100.0 mL with the same acid solution. Dilute 1.0 mL of this solution to 100.0 mL with a 103.0 g/L solution of hydrochloric acid R. Introduce 2.0 mL of this solution into a 20 mL vial, add 1.0 mL of a 126.0 g/L solution of sodium hydroxide R, and close the vial immediately.
Column:
— material: fused silica;
— size: l = 30 m, Ø = 0.53 mm;
— stationary phase: cyanopropyl(3)phenyl(3)methyl(94)polysiloxane R (film thickness 3 μm).
Carrier gas helium for chromatography R.
Flow rate 4.0 mL/min.
Split ratio 1:1.
Static head-space conditions that may be used:
— equilibration temperature: 90 °C;
— equilibration time: 25 min;
— pressurisation time: 2 min.
Temperature:
| Time
(min) |
Temperature
(°C) |
|
| Column
Injection port Detector |
0 – 10
10 – 15 |
130 → 230
230 225 250 |
Detection Flame ionisation.
Retention time Impurity A = about 6 min.
System suitability Reference solution:
— repeatability: maximum relative standard deviation of 15.0 per cent determined on 6 injections.
Calculation of content:
— for impurity A, use the concentration of impurity A in the reference solution.
Limit:
— impurity A: maximum 20 ppm.
Loss on drying (2.2.32)
Maximum 1.0 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.250 g in a mixture of 5.0 mL of 0.01 M hydrochloric acid and 50 mL of ethanol (96 per cent) R. Carry out a potentiometric titration (2.2.20), using 0.1 M sodium hydroxide. Read the volume added between the 2 points of inflexion.
1 mL of 0.1 M sodium hydroxide is equivalent to 28.28 mg of C15H23ClN2O.
IMPURITIES
Specified impurities A.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B, C, D, E.
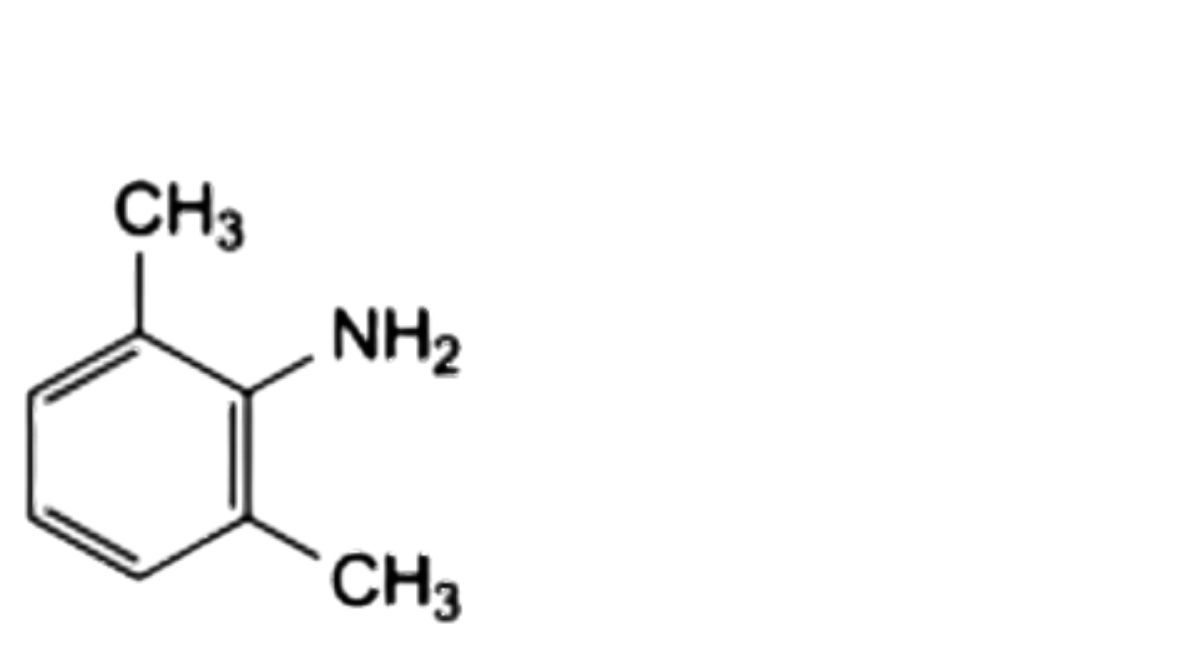
A. 2,6-dimethylaniline,
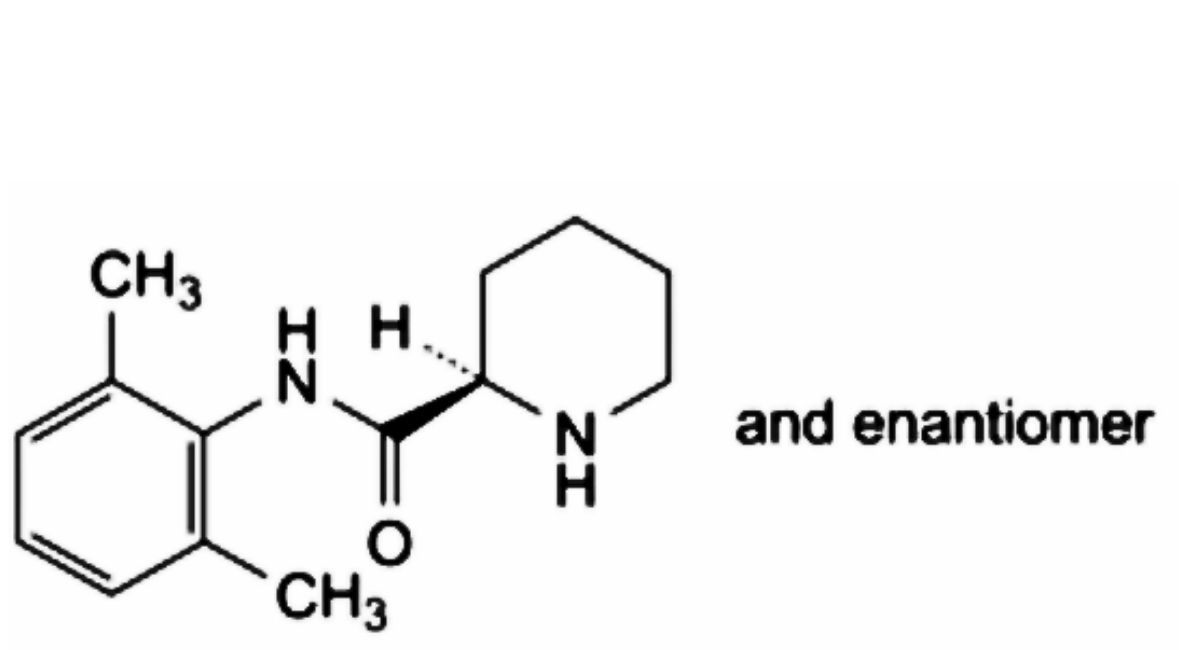
B. (2RS)-N-(2,6-dimethylphenyl)piperidine-2-carboxamide,
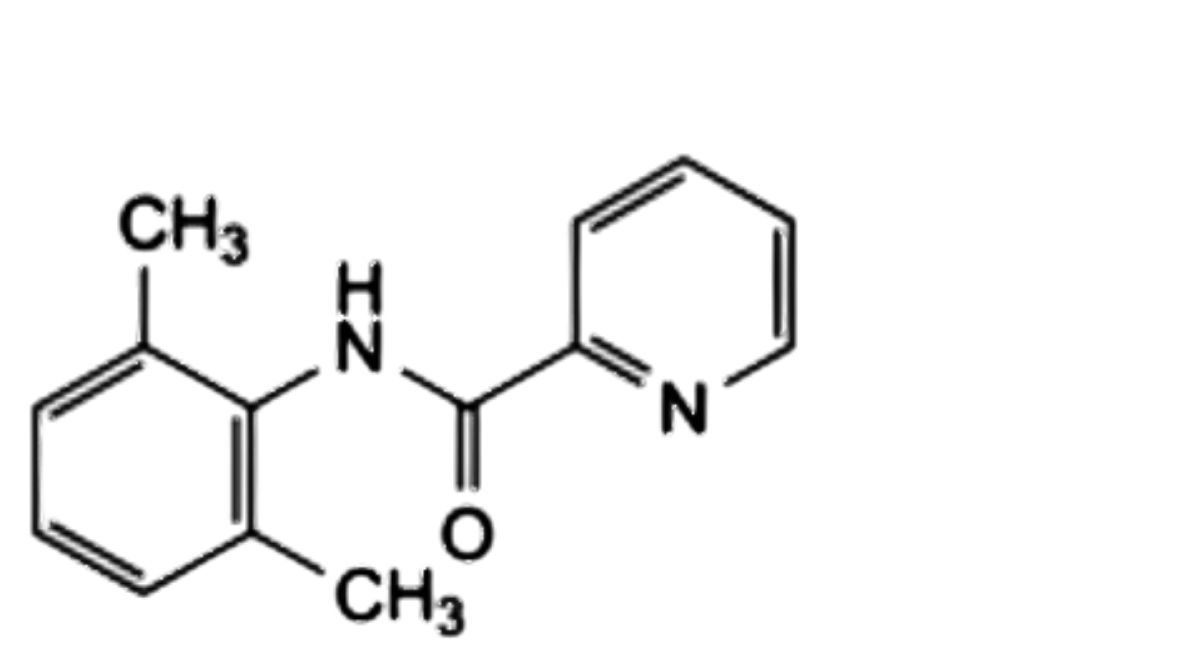
C. N-(2,6-dimethylphenyl)pyridine-2-carboxamide,
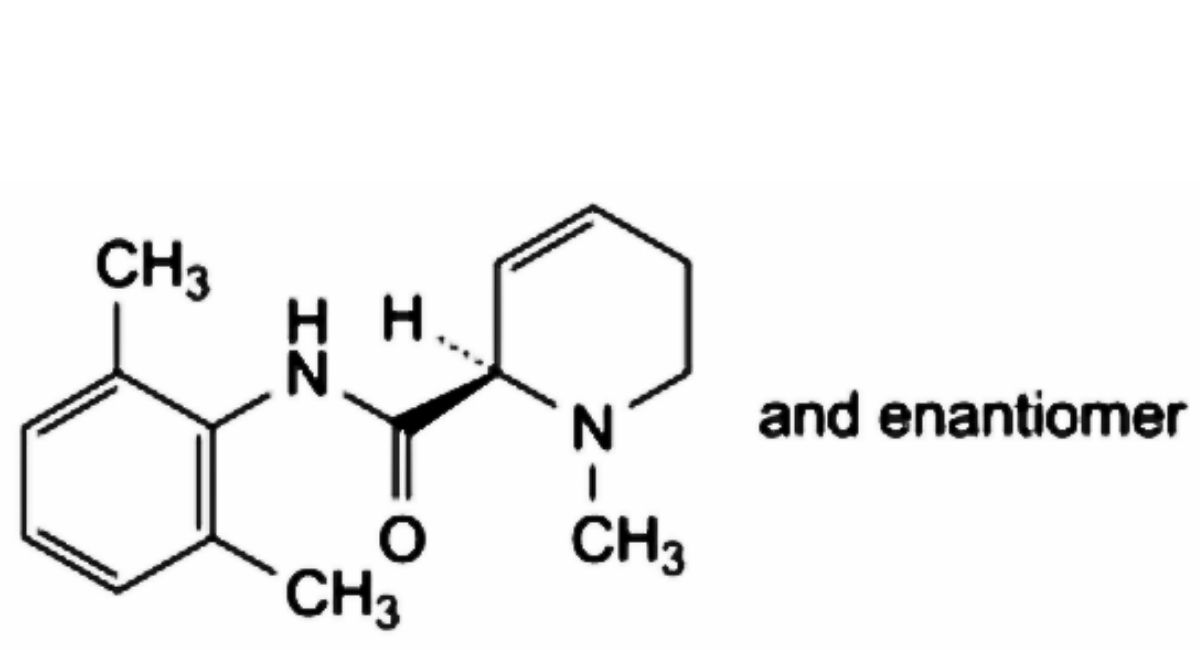
D. (2RS)-N-(2,6-dimethylphenyl)-1-methyl-1,2,5,6-tetrahydropyridine-2-carboxamide,
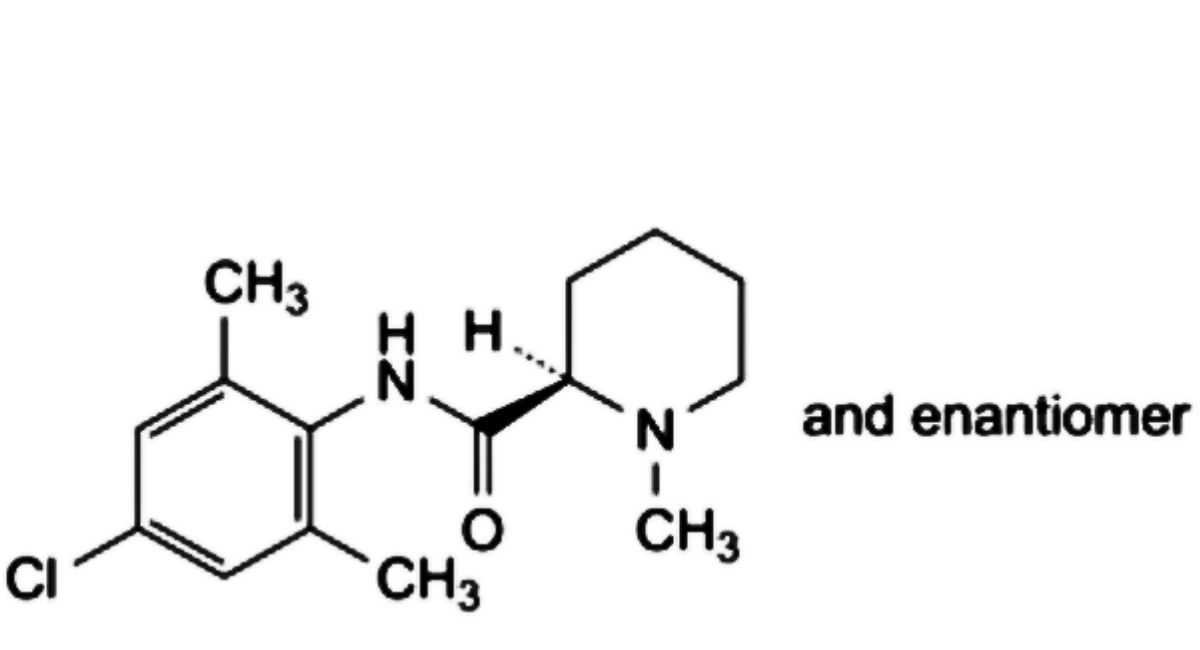
E. (2RS)-N-(4-chloro-2,6-dimethylphenyl)-1-methylpiperidine-2-carboxamide.


