


Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Alpha-adrenoceptor agonist.
Preparation
Xylometazoline Nasal Drops
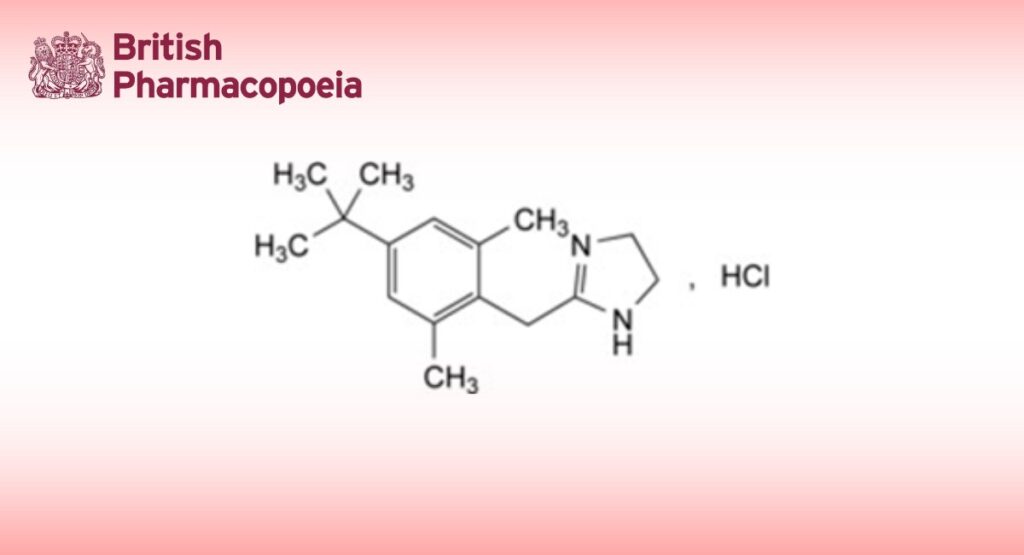
DEFINITION
2-[4-(1,1-Dimethylethyl)-2,6-dimethylbenzyl]-4,5-dihydro-1H-imidazole hydrochloride.
Content
99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Freely soluble in water, in ethanol (96 per cent) and in methanol.
IDENTIFICATION
First identification: A, E.
Second identification: B, C, D, E.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison xylometazoline hydrochloride CRS.
B. Thin-layer chromatography (2.2.27).
Test solution Dissolve 20 mg of the substance to be examined in methanol R and dilute to 5 mL with the same solvent.
Reference solution Dissolve 20 mg of xylometazoline hydrochloride CRS in methanol R and dilute to 5 mL with the same solvent.
Plate TLC silica gel G plate R.
Mobile phase concentrated ammonia R, methanol R (5:100 V/V). Application 5 µL.
Development Over 2/3 of the plate.
Drying In air.
Chlorine treatment At the bottom of a chromatographic tank place a beaker containing a mixture of 1 volume of hydrochloric acid R1, 1 volume of water R and 2 volumes of a 15 g/L solution of potassium permanganate R. Close the tank and allow to stand for 15 min. Place the dried plate in the tank and reclose the tank. Leave the plate in contact with the chlorine vapour for 5 min. Withdraw the plate and place it in a current of cold air until the excess of chlorine is removed and an area of the coating below the points of application does not give a blue colour with a drop of potassium iodide and starch solution R.
Detection Spray with potassium iodide and starch solution R.
Results The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with the reference solution.
C. Dissolve about 0.5 mg in 1 mL of methanol R. Add 0.5 mL of a freshly prepared 50 g/L solution of sodium nitroprusside R and 0.5 mL of a 20 g/L solution of sodium hydroxide R. Allow to stand for 10 min and add 1 mL of an 80 g/L solution of sodium hydrogen carbonate R. A violet colour develops.
D. Dissolve 0.2 g in 1 mL of water R, add 2.5 mL of ethanol (96 per cent) R and 2 mL of 1 M sodium hydroxide. Mix thoroughly and examine in ultraviolet light at 365 nm. The solution shows no fluorescence or at most the same fluorescence as a blank solution prepared in the same manner. The identification is not valid unless a solution prepared in the same manner using naphazoline hydrochloride CRS instead of the substance to be examined shows a distinct bluish fluorescence.
E. It gives reaction (a) of chlorides (2.3.1).
TESTS
Appearance of solution
The solution is clear (2.2.1) and not more intensely coloured than reference solution Y6 (2.2.2, Method II). Dissolve 2.5 g in water R and dilute to 50.0 mL with the same solvent.
Acidity or alkalinity
Dissolve 0.25 g in carbon dioxide-free water R and dilute to 25 mL with the same solvent. Add 0.1 mL of methyl red solution R and 0.1 mL of 0.01 M hydrochloric acid.
The solution is red. Not more than 0.2 mL of 0.01 M sodium hydroxide is required to change the colour of the indicator to yellow.
Related substances
Liquid chromatography (2.2.29).
Test solution Dissolve 50.0 mg of the substance to be examined in water R and dilute to 50.0 mL with the same solvent. Allow to stand for 1 h before injection.
Reference solution (a) Dilute 5.0 mL of the test solution to 100.0 mL with water R. Dilute 2.0 mL of this solution to 100.0 mL with water R.
Reference solution (b) Dissolve 5.0 mg of xylometazoline impurity A CRS and 5 mg of the substance to be examined in water R and dilute to 50.0 mL with the same solvent. Dilute 10.0 mL of this solution to 50.0 mL with water R. Reference solution (c) Dilute 5.0 mL of reference solution (b) to 50.0 mL with water R.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: end-capped octadecylsilyl silica gel for chromatography with embedded polar groups R (5 µm).
Mobile phase:
— mobile phase A: 1.36 g/L solution of potassium dihydrogen phosphate R adjusted to pH 3.0 with phosphoric acid R;
— mobile phase B: acetonitrile for chromatography R;
| Time (min) | Mobile phase A (per cent V/V) | Mobile phase B (per cent V/V) |
| 0 – 5 | 70 | 30 |
| 5 – 20 | 70 → 15 | 30 → 85 |
| 20 – 35 | 15 | 85 |
Flow rate 1.0 mL/min.
Detection Spectrophotometer at 220 nm.
Injection 10 µL.
Relative retention With reference to xylometazoline (retention time = about 7.2 min): impurity A = about 0.79.
System suitability Reference solution (b):
— resolution: minimum 2.5 between the peaks due to impurity A and xylometazoline.
Limits:
— impurity A: not more than the area of the corresponding peak in the chromatogram obtained with reference solution (c) (0.2 per cent);
— unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (a) (0.10 per cent);
— total: not more than 5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.5 per cent);
— disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.05 per cent).
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.200 g in 25 mL of anhydrous acetic acid R and add 10 mL of acetic anhydride R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 28.08 mg of C16H25ClN2.
STORAGE
Protected from light.
IMPURITIES
Specified impurities A.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B, C, D, E, F.
A. N-(2-aminoethyl)-2-[4-(1,1-dimethylethyl)-2,6-dimethylphenyl]acetamide,
B. 2-(chloromethyl)-5-(1,1-dimethylethyl)-1,3-dimethylbenzene,
C. [4-(1,1-dimethylethyl)-2,6-dimethylphenyl]acetonitrile,
D. 1-(1,1-dimethylethyl)-3,5-dimethylbenzene,
E. ethane-1,2-diamine mono(4-methylbenzenesulfonate),
F. [4-(1,1-dimethylethyl)-2,6-dimethylphenyl]acetic acid.


