


Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Vinca alkaloid cytotoxic.
DEFINITION
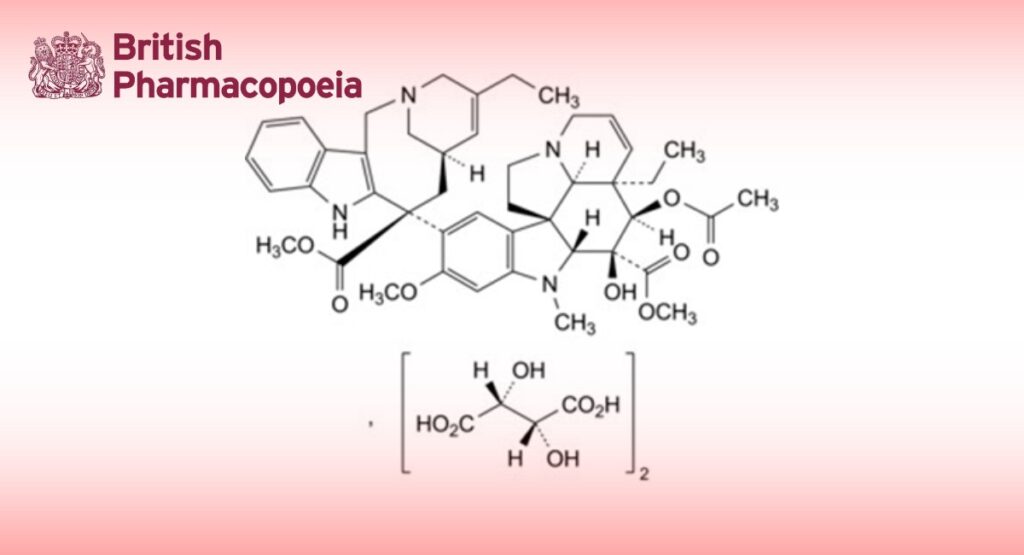
4′-Deoxy-3′,4′-didehydro-8′-norvincaleukoblastine dihydrogen bis[(2R,3R)-2,3-dihydroxybutanedioate].
Content
98.0 per cent to 102.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white powder, hygroscopic.
Solubility
Freely soluble in water and in methanol, practically insoluble in hexane.
IDENTIFICATION
A. Infrared absorption spectrophotometry (2.2.24).
Preparation Dissolve 10 mg in 5 mL of water R. Add 0.5 mL of sodium hydroxide solution R. Extract with 5 mL of methylene chloride R. Dry the organic layer over anhydrous sodium sulfate R, filter and reduce its volume to about 0.5 mL by evaporation and apply to a disc of potassium bromide R. Evaporate and record the spectrum.
Comparison vinorelbine tartrate CRS, treated as described above.
B. It gives reaction (b) of tartrates (2.3.1).
TESTS
Solution S
Dissolve a quantity equivalent to 0.140 g of the anhydrous substance in carbon dioxide-free water R and dilute to 10.0 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and its absorbance (2.2.25) at 420 nm is not greater than 0.030.
pH (2.2.3)
3.3 to 3.8 for solution S.
Related substances
Liquid chromatography (2.2.29): use the normalisation procedure.
Solvent mixture Mobile phase A, mobile phase B (45:55 V/V).
Test solution Dissolve 35.0 mg of the substance to be examined in the solvent mixture and dilute to 25.0 mL with the solvent mixture.
Reference solution (a) Dissolve 14 mg of vinorelbine tartrate CRS in water R and dilute to 10 mL with the same solvent. Expose this solution for 1 h to a xenon lamp apparatus at a wavelength of 310-880 nm, supplying a dose of 1600 kJ/m2 at a fluence rate of 500 W/m2 in order to generate impurity A.
Reference solution (b) Dilute 1.0 mL of the test solution to 20.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 100.0 mL with the solvent mixture.
Reference solution (c) Dissolve 7 mg of vinorelbine for peak identification A CRS (containing impurity K) in the solvent mixture and dilute to 5 mL with the solvent mixture.
Reference solution (d) Dissolve 7 mg of vinorelbine for peak identification B CRS (containing impurities C and I) in the solvent mixture and dilute to 5 mL with the solvent mixture.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: spherical end-capped octadecylsilyl silica gel for chromatography R (5 µm);
— temperature: 30 °C.
Mobile phase:
— mobile phase A: mix 14 mL of diethylamine R and 986 mL of water for chromatography R and adjust to pH 7.5 with dilute phosphoric acid R;
— mobile phase B: acetonitrile R, methanol R (20:80 V/V);
| Time (min) | Mobile phase A (per cent V/V) | Mobile phase B (per cent V/V) |
| 0 – 5 | 45 | 55 |
| 5 – 45 | 45 → 20 | 55 → 80 |
| 45 – 50 | 20 | 80 |
Flow rate 2.0 mL/min.
Detection Spectrophotometer at 267 nm.
Autosampler Set at 5 °C.
Injection 20 µL.
Identification of impurities Use the chromatogram obtained with reference solution (a) to identify the peak due to impurity A; use the chromatogram supplied with vinorelbine for peak identification B CRS and the chromatogram obtained with reference solution (d) to identify the peaks due to impurities C and I; use the chromatogram supplied with vinorelbine for peak identification A CRS and the chromatogram obtained with reference solution (c) to identify the peak due to impurity K.
Relative retention With reference to vinorelbine (retention time = about 20 min): impurity C = about 0.65; impurity K = about 0.8; impurity A = about 0.9; impurity I = about 1.2.
System suitability Reference solution (a):
— resolution: minimum 1.5 between the peaks due to impurity A and vinorelbine.
Limits:
— impurity A: maximum 0.3 per cent;
— impurities C, I, K: for each impurity, maximum 0.2 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 0.5 per cent;
— reporting threshold: 0.05 per cent (principal peak in the chromatogram obtained with reference solution (b)).
Boron
Maximum 50 ppm.
Test solution Dissolve 0.10 g of the substance to be examined in 2 mL of water R. Slowly add 10.0 mL of sulfuric acid R while cooling in iced water. Stir and allow to warm to room temperature. Add 10.0 mL of a 0.5 g/L solution of carminic acid R in sulfuric acid R.
Reference solution Dilute 2.5 mL of a 0.572 g/L solution of boric acid R to 100.0 mL with water R. To 2.0 mL of this solution slowly add 10.0 mL of sulfuric acid R while cooling in iced water. Stir and allow to warm to room temperature. Add 10.0 mL of a 0.5 g/L solution of carminic acid R in sulfuric acid R.
Blank solution To 2.0 mL of water R slowly add 10.0 mL of sulfuric acid R while cooling in iced water. Stir and allow to warm to room temperature. Add 10.0 mL of a 0.5 g/L solution of carminic acid R in sulfuric acid R.
After 45 min, measure the absorbance (2.2.25) of the test solution and the reference solution, between 560 nm and 650 nm, using the blank solution as compensation liquid. The maximum absorbance value of the test solution is not greater than that of the reference solution.
Fluorides
Maximum 50 ppm.
Potentiometry (2.2.36, Method I) using a fluoride-selective indicator electrode and a silver-silver chloride reference electrode.
Test solution Dissolve 0.19 g of the substance to be examined in 20 mL of water R. Add 5.0 mL of total-ionic-strength- adjustment buffer R and dilute to 50 mL with water R.
Reference solutions To 0.6 mL, 0.8 mL, 1.0 mL, 1.2 mL and 1.4 mL of fluoride standard solution (10 ppm F) R, add 5.0 mL of total-ionic-strength-adjustment buffer R and dilute to 50 mL with water R.
Introduce the electrodes into the reference solutions and allow to stand for 5 min. Determine the potential difference between the electrodes after 1 min of stabilisation. Using semi-logarithmic paper plot the potential difference obtained for each reference solution as a function of concentration of fluoride. Using exactly the same conditions, determine the potential difference obtained with the test solution and calculate the content of fluoride.
Water (2.5.12)
Maximum 4.0 per cent, determined on 0.250 g.
ASSAY
Dissolve 0.350 g in 40 mL of glacial acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 53.96 mg of C53H66N4O20.
STORAGE
Under an inert gas, protected from light, at a temperature not exceeding -15 °C.
IMPURITIES
Specified impurities A, C, I, K.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B, D, E, F, G, H, J.
A. 3,4′-dideoxy-3,6ξ-epoxy-3′,4′,7,8-tetradehydro-6,7-dihydro-8′-nor-3ξ-vincaleukoblastine,
B. O4-deacetyl-4′-deoxy-3′,4′-didehydro-8′-norvincaleukoblastine,
C. unknown structure,
D. (6′RS)-4′-deoxy-3′,4′-didehydro-8′-norvincaleukoblastine 6′-oxide,
E. 4′-deoxy-3′α,4′α-epoxyvincaleukoblastine (leurosine),
F. (6′RS)-4′-deoxy-6′-methyl-3′,4′-didehydro-6′-azonia-8′-norvincaleukoblastine,
G. 4′-deoxy-3′α,4′α-epoxy-8′-norvincaleukoblastine,
H. 4′-deoxy-3′,4′-didehydro-8′,24-dinorvincaleukoblastine,
I. 17-bromo-4′-deoxy-3′,4′-didehydro-8′-norvincaleukoblastine,
J. 4′-deoxy-3′,4′-didehydrovincaleukoblastine,
K. 4′-deoxy-8′-nor-4′αβ-vincaleukoblastine.


