


Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Purine nucleoside analogue; antiviral (herpesviruses).
Preparation
Valaciclovir Tablets
DEFINITION
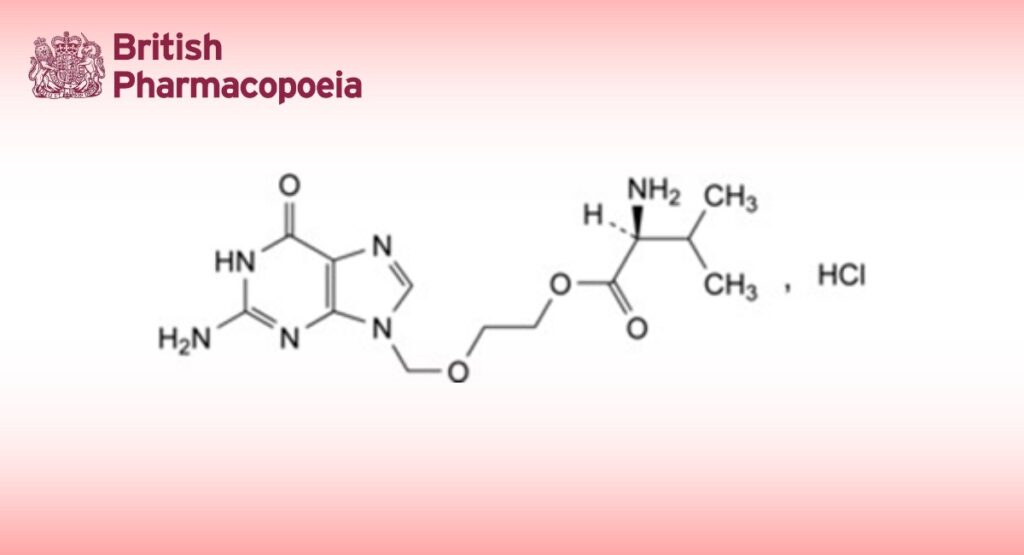
2-[(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl L-valinate hydrochloride.
Content
95.0 per cent to 102.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white powder.
Solubility
Freely soluble in water, slightly soluble in anhydrous ethanol. It shows polymorphism (5.9).
IDENTIFICATION
Carry out either tests A, B, C, E or tests A, B, D, E.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison anhydrous valaciclovir hydrochloride CRS.
If the spectra obtained in the solid state show differences, dissolve the substance to be examined and the reference substance separately in the minimum volume of anhydrous ethanol R and evaporate to dryness in a desiccator, under high vacuum, over diphosphorus pentoxide R. Record new spectra using the residues.
B. It gives reaction (a) of chlorides (2.3.1).
C. It complies with the limit for impurity R given in test A for related substances.
D. Optical rotation (2.2.7): laevorotatory.
Dissolve 2.50 g in water R and dilute to 50.0 mL with the same solvent.
E. Water (see Tests).
TESTS
Impurities E, F and G
Thin-layer chromatography (2.2.27).
Test solution Dissolve 0.250 g of the substance to be examined in 2 mL of water R and dilute to 5.0 mL with ethanol (96 per cent) R.
Reference solution (a) Dissolve 5 mg of valaciclovir impurity D CRS, 5.0 mg of valaciclovir impurity E CRS, 5.0 mg of valaciclovir impurity G CRS and 8.4 mg of valaciclovir impurity F para-toluenesulfonate CRS in a mixture of 2 mL of water R and 6 mL of ethanol (96 per cent) R, and dilute to 10.0 mL with ethanol (96 per cent) R.
Reference solution (b) Dilute 3.0 mL of reference solution (a) to 10.0 mL with ethanol (96 per cent) R. Reference solution (c) Dilute 2.0 mL of reference solution (a) to 10.0 mL with ethanol (96 per cent) R. Reference solution (d) Dilute 0.5 mL of reference solution (a) to 10.0 mL with ethanol (96 per cent) R. Plate TLC silica gel F254 plate R (2-10 µm).
Pretreatment Wash the plate with methanol R until the solvent front has migrated over at least 4/5 of the plate; allow the plate to dry.
Mobile phase concentrated ammonia R, tetrahydrofuran R, methanol R, methylene chloride R (3:12:34:54 V/V/V/V); use freshly prepared mobile phase.
Application 4 µL of the test solution and reference solutions (b), (c) and (d).
Development Over 4/5 of the plate.
Drying In a current of air.
Detection Examine in ultraviolet light at 254 nm for impurities E and G; spray with a 0.1 g/L solution of fluorescamine R in ethylene chloride R and examine in ultraviolet light at 365 nm for impurity F.
Retardation factors Impurity A = about 0; impurity B = about 0.2; valaciclovir = about 0.3; impurity C = about 0.5; impurity D = about 0.6; impurity E = about 0.7; impurity F = about 0.75; impurity G = about 0.79; impurity C is masked by the leading edge of the spot due to valaciclovir; impurities F and G may co-elute, but this does not adversely affect their quantification because they are visualised differently.
System suitability The chromatograms obtained with reference solutions (b), (c) and (d) each show 3 clearly separated spots when examined under ultraviolet light at 254 nm, due to impurities D, E and G.
Limits:
— impurity E: any spot due to impurity E is not more intense than the corresponding spot in the chromatogram obtained with reference solution (c) (0.2 per cent);
— impurity F: any spot due to impurity F is not more intense than the corresponding spot in the chromatogram obtained with reference solution (b) (0.3 per cent calculated as hydrochloride salt);
— impurity G: any spot due to impurity G is not more intense than the corresponding spot in the chromatogram obtained with reference solution (d) (0.05 per cent).
Related substances
A. Impurities A, B, I and R.
Liquid chromatography (2.2.29): use the normalisation procedure.
Test solution Dissolve 50.0 mg of the substance to be examined in a 0.5 per cent V/V solution of hydrochloric acid R and dilute to 100.0 mL with the same solution.
Reference solution (a) Dissolve 2.5 mg of valaciclovir for system suitability CRS (containing impurities A, B, C, D, H, I, J, M and R) in a 0.5 per cent V/V solution of hydrochloric acid R and dilute to 5.0 mL with the same solution.
Reference solution (b) Dissolve 50.0 mg of anhydrous valaciclovir hydrochloride CRS in a 0.5 per cent V/V solution of hydrochloric acid R and dilute to 100.0 mL with the same solution.
Reference solution (c) Dilute 3.0 mL of the test solution to 100.0 mL with a 0.5 per cent V/V solution of hydrochloric acid R. Dilute 1.0 mL of this solution to 100.0 mL with a 0.5 per cent V/V solution of hydrochloric acid R.
Column:
— size: l = 0.15 m, Ø = 4.0 mm;
— stationary phase: crown-ether silica gel for chiral separation R (5 µm);
— temperature: 10 °C.
Mobile phase perchloric acid R, methanol R, water for chromatography R (0.5:5:95 V/V/V). Flow rate 0.75 mL/min.
Detection Spectrophotometer at 254 nm.
Injection 10 µL of the test solution and reference solutions (a) and (c).
Run time 1.5 times the retention time of valaciclovir.
Identification of impurities Use the chromatogram supplied with valaciclovir for system suitability CRS and the chromatogram obtained with reference solution (a) to identify the peaks due to impurities A + B, C + R, D, I and M.
Relative retention With reference to valaciclovir (retention time = about 21 min): impurities A and B = about 0.2; impurity I = about 0.4; impurities C and R = about 0.6; impurity D = about 0.7; impurity M = about 1.3.
System suitability Reference solution (a):
— peak-to-valley ratio: minimum 1.5, where Hp = height above the baseline of the peak due to impurity D and
Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to impurities C and R.
Limits:
— correction factor: for the calculation of content, multiply the peak area of impurities A and B by 0.7;
— impurity R: maximum 3.0 per cent; for the calculation, subtract the content of impurity C as determined in test B for related substances from the content of the coeluting impurities C and R as determined in this test;
— sum of impurities A and B: maximum 2.0 per cent;
— impurity I: maximum 0.2 per cent;
— disregard limit: the area of the principal peak in the chromatogram obtained with reference solution (c) (0.03 per cent); disregard any peaks due to impurities other than A + B, C + R or I.
B. Liquid chromatography (2.2.29): use the normalisation procedure. Use the solutions within 24 h of preparation.
Solvent mixture ethanol (96 per cent) R, water R (20:80 V/V).
Test solution Dissolve 40 mg of the substance to be examined in the solvent mixture and dilute to 100.0 mL with the solvent mixture.
Reference solution (a) Dissolve 2.5 mg of valaciclovir for system suitability CRS (containing impurities A, B, C, D, H, I, J, M and R) in the solvent mixture and dilute to 5.0 mL with the solvent mixture.
Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 20.0 mL with the solvent mixture.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: end-capped phenylhexylsilyl silica gel for chromatography R (5 µm);
— temperature: 15 °C.
Mobile phase:
— mobile phase A: trifluoroacetic acid R, water for chromatography R (0.2:100 V/V);
— mobile phase B: trifluoroacetic acid R, methanol R2 (0.2:100 V/V);
| Time (min) | Mobile phase A (per cent V/V) | Mobile phase B (per cent V/V) |
| 0 – 5 | 90 | 10 |
| 5 – 35 | 90 → 60 | 10 → 40 |
Flow rate 0.8 mL/min.
Detection Spectrophotometer at 254 nm.
Injection 10 µL.
Identification of impurities Use the chromatogram supplied with valaciclovir for system suitability CRS and the chromatogram obtained with reference solution (a) to identify the peaks due to impurities A, B, C, D, H, I, J and M.
Relative retention With reference to valaciclovir (retention time = about 19 min): impurity A = about 0.3; impurity B = about 0.4; impurity H = about 0.5; impurity C = about 1.06; impurity I = about 1.09; impurity D = about 1.2; impurity J = about 1.3; impurity M = about 1.6.
System suitability Reference solution (a):
— peak-to-valley ratio: minimum 2.5, where Hp = height above the baseline of the peak due to impurity C and
Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to valaciclovir;
— the chromatogram obtained is similar to the chromatogram supplied with valaciclovir for system suitability CRS. Limits:
— impurity M: maximum 1.5 per cent;
— impurity D: maximum 0.5 per cent;
— impurity C: maximum 0.3 per cent;
— impurities H, J: for each impurity, maximum 0.10 per cent;
— unspecified impurities: for each impurity, maximum 0.05 per cent;
— disregard limit: 0.6 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.03 per cent); disregard the peaks due to impurities A, B and I.
Limit:
— total for tests A and B: maximum 5.0 per cent.
Chloride
9.4 to 9.9 per cent (anhydrous and solvent-free substance).
Dissolve 0.350 g in 100 mL of water R and add 0.2 mL of nitric acid R. Carry out a potentiometric titration (2.2.20), using 0.1 M silver nitrate. Use a silver indicator electrode and a silver-silver chloride reference electrode or a combined silver electrode. Discard the result from the first titration, which is used to condition the electrodes. Carry out a blank titration.
1 mL of 0.1 M silver nitrate is equivalent to 3.543 mg of Cl.
Water (2.5.12)
Maximum 2.0 per cent, determined on 0.250 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Liquid chromatography (2.2.29) as described in test A for related substances with the following modification.
Injection Test solution and reference solution (b).
Calculate the percentage content of C13H21ClN6O4 taking into account the assigned content of anhydrous valaciclovir hydrochloride CRS.
IMPURITIES
Specified impurities A, B, C, D, E, F, G, H, I, J, M, R.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) K, L, N, O, P, Q.
A. 2-amino-1,9-dihydro-6H-purin-6-one (guanine),
B. 2-amino-9-[(2-hydroxyethoxy)methyl]-1,9-dihydro-6H-purin-6-one (aciclovir),
C. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl N-methyl-L-valinate,
D. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl N-ethyl-L-valinate,
E. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl N-[(benzyloxy)carbonyl]-L-valinate,
F. 2-hydroxyethyl L-valinate,
G. N,N-dimethylpyridin-4-amine,
H. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl L-alaninate,
I. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl acetate,
J. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl L-isoleucinate,
K. 9-[(2-hydroxyethoxy)methyl]-2-[[[(6-oxo-6,9-dihydro-1H-purin-2-yl)amino]methyl]amino]-1,9-dihydro-6H-purin-6-one,
L. 2,2′-(methylenediazanediyl)bis[9-[(2-hydroxyethoxy)methyl]-1,9-dihydro-6H-purin-6-one],
M. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl N-formyl-L-valinate,
N. 2-[[6-oxo-2-[[[(6-oxo-6,9-dihydro-1H-purin-2-yl)amino]methyl]amino]-1,6-dihydro-9H-purin-9-yl]methoxy]ethyl L- valinate,
O. 2-[[2-[[[[9-[(2-hydroxyethoxy)methyl]-6-oxo-6,9-dihydro-1H-purin-2-yl]amino]methyl]amino]-6-oxo-1,6-dihydro-9H- purin-9-yl]methoxy]ethyl L-valinate,
P. [methylenebis[azanediyl(6-oxo-1,6-dihydro-9H-purine-2,9-diyl)methyleneoxyethan-2,1-diyl]] di-L-valinate,
Q. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl N-[[(6-oxo-6,9-dihydro-1H-purin-2-yl)amino]methyl]-L- valinate,
R. 2-[(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy]ethyl D-valinate.


