



Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Immunomodulator; treatment of multiple sclerosis.
Ph Eur
DEFINITION
Tablets containing Teriflunomide (3036), for human use.
They comply with the monograph Tablets (0478) and the following additional requirements.
Content
95.0 per cent to 105.0 per cent of the content of teriflunomide (C12H9F3N2O2) stated on the label.
IDENTIFICATION
A. Record the UV spectrum of the principal peak in the chromatograms obtained with the solutions used in the assay, with a diode array detector, in the range of 200-400 nm.
Results The UV spectrum of the principal peak in the chromatogram obtained with the test solution is similar to the UV spectrum of the principal peak in the chromatogram obtained with reference solution (d).
B. Examine the chromatograms obtained in the assay.
Results The principal peak in the chromatogram obtained with the test solution is similar in retention time and size to the principal peak in the chromatogram obtained with reference solution (d).
TESTS
Related substances
Liquid chromatography (2.2.29).
Buffer solution 3.85 g/L solution of ammonium acetate R adjusted to pH 5.5 with glacial acetic acid R. Solvent mixture Buffer solution, acetonitrile R (20:80 V/V).
Test solution To 10 tablets, add a volume of the solvent mixture equivalent to about half of the final volume. Dissolve the tablets by shaking for approximately 20 min and sonicating for about 3 min. Dilute to volume with the solvent mixture to obtain a concentration of teriflunomide of 0.7 mg/mL and filter. Dilute 3.0 mL of the filtrate to 10.0 mL with the solvent mixture.
Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with the solvent mixture.
Reference solution (b) Dissolve 10 mg of leflunomide impurity A CRS (teriflunomide impurity A) in acetonitrile R and dilute to 250 mL with the same solvent. Dilute 1 mL of the solution to 200 mL with the solvent mixture.
Reference solution (c) Dissolve 5 mg of teriflunomide impurity B CRS in acetonitrile R and dilute to 100 mL with the same solvent. Dilute 2 mL of the solution to 50 mL with the solvent mixture. To 1 mL of this solution add 1 mL of reference solution (b) and dilute to 10 mL with the solvent mixture.
Reference solution (d) Dissolve 21.0 mg of teriflunomide for assay CRS in 40 mL of the solvent mixture and dilute to
100.0 mL with the solvent mixture.
Reference solution (e) Dilute 1.0 mL of reference solution (a) to 10.0 mL with the solvent mixture.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: end-capped solid core octadecylsilyl silica gel for chromatography R (2.7 µm);
— temperature: 40 °C.
Mobile phase:
— mobile phase A: acetonitrile for chromatography R, buffer solution (10:90 V/V);
— mobile phase B: buffer solution, acetonitrile for chromatography R (10:90 V/V);
| Time (min) | Mobile phase A (per cent V/V) | Mobile phase B (per cent V/V) |
| 0-2 | 76 | 24 |
| 2-12 | 76 → 23 | 24 → 77 |
Flow rate 1.0 mL/min.
Detection Spectrophotometer at 249 nm.
Injection 5 µL of the test solution and reference solutions (c) and (e).
Identification of impurities Use the chromatogram obtained with reference solution (c) to identify the peaks due to impurities A and B.
Relative retention With reference to teriflunomide (retention time = about 5 min): impurity B = about 1.5; impurity A = about 1.6.
System suitability Reference solution (c):
— resolution: minimum 1.5 between the peaks due to impurities B and A;
— signal-to-noise ratio: minimum 10 for the peak due to impurity A.
Calculation of percentage contents:
— for each impurity, use the concentration of teriflunomide in reference solution (e).
Limits:
— impurity A: maximum 0.01 per cent;
— impurity B: maximum 0.30 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 0.40 per cent;
— reporting threshold: 0.05 per cent; do not disregard the peak due to impurity A.
Dissolution1 (2.9.3, Apparatus 2).
Dissolution medium 6.8 g/L solution of potassium dihydrogen phosphate R adjusted to pH 6.8 with a 200 g/L solution of sodium hydroxide R. Use 1000 mL of the medium.
Rotation speed 50 r/min.
Time 30 min.
Analysis Ultraviolet and visible absorption spectrophotometry (2.2.25).
Test solution Samples withdrawn from the dissolution vessel and filtered. Measure the absorbance of the solutions at the absorption maximum at 292 nm.
Calculate the amount of dissolved teriflunomide (C12H9F3N2O2), expressed as a percentage of the content stated on the label, taking the specific absorbance to be 854.
Acceptance criterion:
— Q = 75 per cent after 30 min.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modifications.
Injection Test solution and reference solution (d).
System suitability Reference solution (d):
— symmetry factor: maximum 2.0 for the peak due to teriflunomide;
— repeatability: maximum relative standard deviation of 1.5 per cent determined on 6 injections.
Calculate the percentage content of teriflunomide (C12H9F3N2O2) taking into account the assigned content of teriflunomide for assay CRS.
IMPURITIES
Specified impurities A, B.
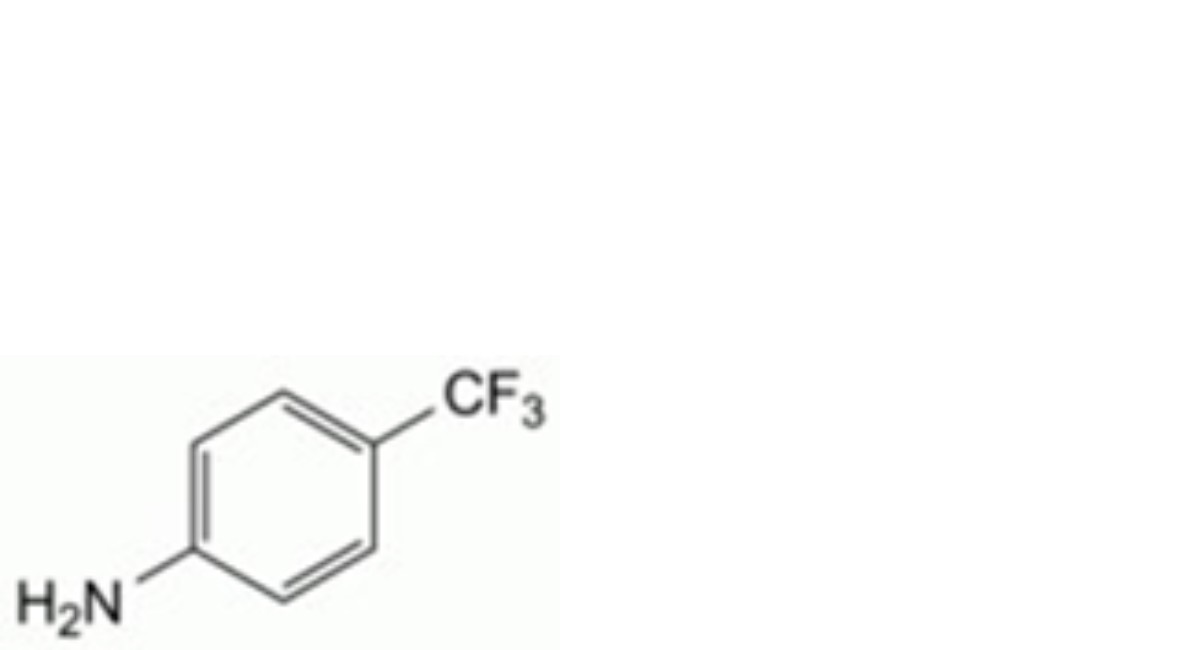
A. 4-(trifluoromethyl)aniline (leflunomide impurity A),
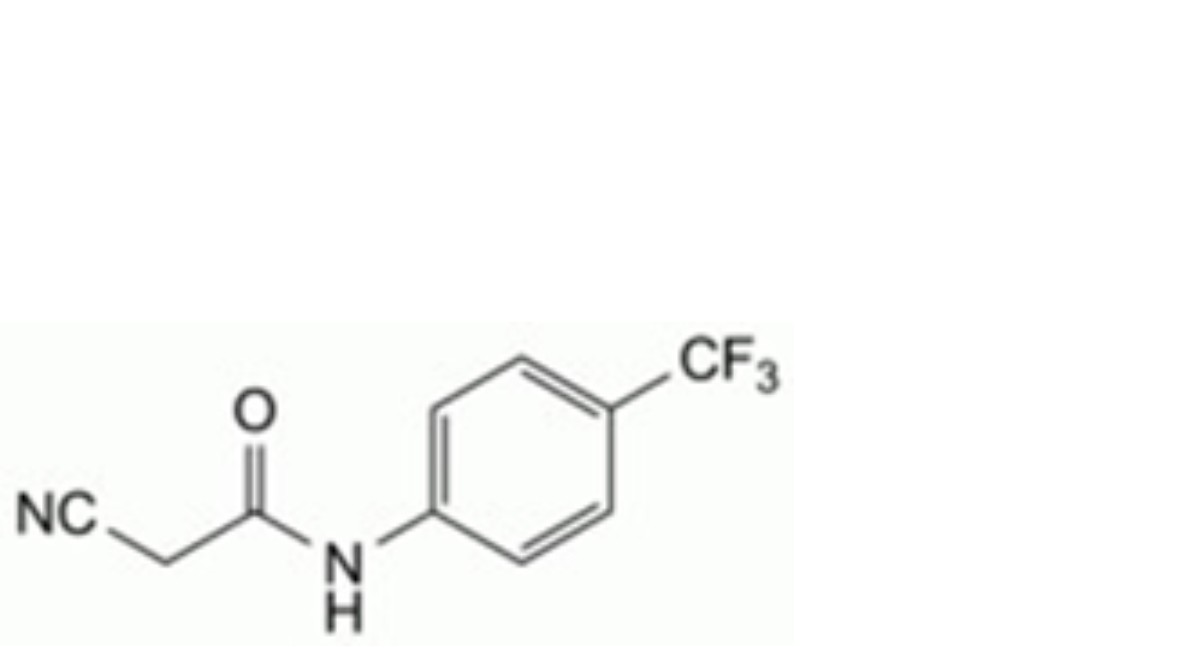
B. 2-cyano-N-[4-(trifluoromethyl)phenyl]acetamide.
Ph Eur
1 The test approved in the marketing authorisation is to be used for routine quality control to confirm batch-to-batch consistency. For more information please consult Ph. Eur. 1. General Notices.


