



BP 2025 (Ph. Eur. 11.6 update)
(Ph. Eur. monograph 1046) 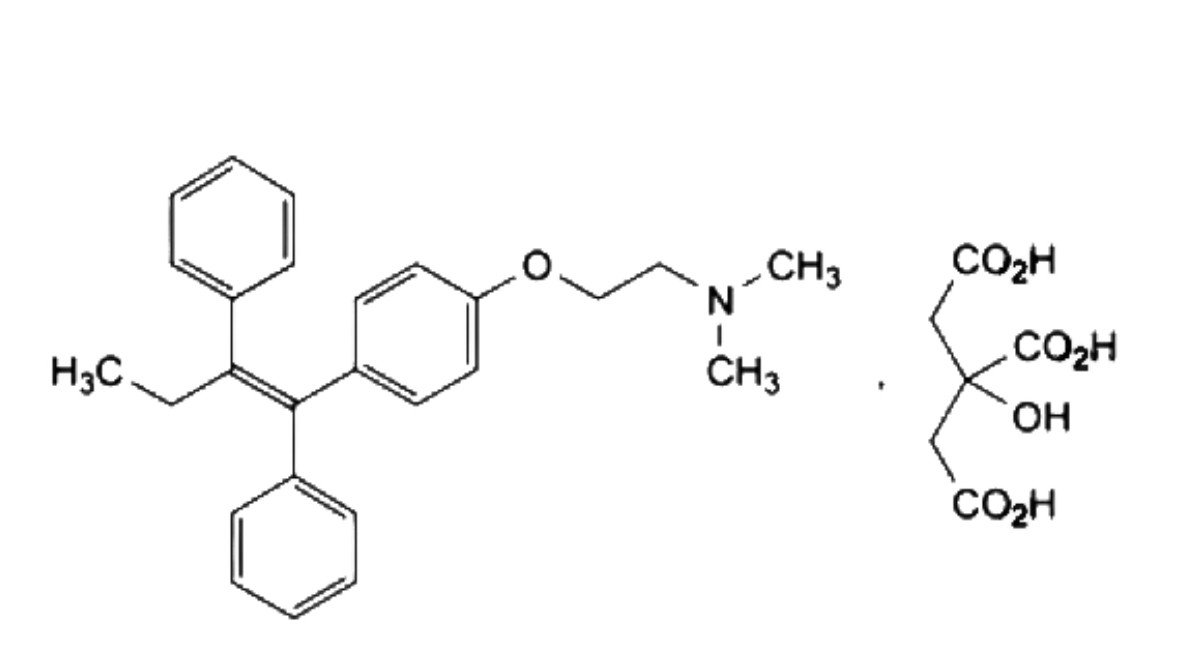
C32H37NO8 563.6 54965-24-1
Action and use
Selective estrogen receptor modulator.
Preparations
Tamoxifen Oral Solution
Tamoxifen Tablets
Ph Eur
DEFINITION
2-[4-[(1Z)-1,2-Diphenylbut-1-en-1-yl]phenoxy]-W,W-dimethylethan-1-amine dihydrogen 2-hydroxypropane-1,2,3- tricarboxylate.
Content
99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Slightly soluble in water, soluble in methanol, slightly soluble in acetone.
It shows polymorphism (5.9).
IDENTIFICATION
First identification: A.
Second identification: B.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison tamoxifen citrate CRS.
If the spectra obtained in the solid state show differences, dissolve the substance to be examined and the reference substance separately in acetone R, evaporate to dryness and record new spectra using the residues.
B. Thin-layer chromatography (2.2.27).
Test solution Dissolve 5 mg of the substance to be examined in methanol R and dilute to 5.0 mL with the same solvent.
Reference solution Dissolve 5 mg of tamoxifen citrate CRS in methanol R and dilute to 5.0 mL with the same solvent.
Plate TLC silica gel F254 plate R.
Mobile phase concentrated ammonia R, propanol R, toluene R (1:29:70 V/V/V).
Application 5 µL.
Development Over 3/4 of the plate.
Drying At 100 °C for 5 min.
Detection A Examine in ultraviolet light at 254 nm.
Results A The principal spot in the chromatogram obtained with the test solution is similar in position and size to the principal spot in the chromatogram obtained with the reference solution.
Detection B Spray with dilute potassium iodobismuthate solution R.
Results B The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with the reference solution.
TESTS
Related substances
Liquid chromatography (2.2.29). Prepare the solutions immediately before use and protect from light.
Test solution Dissolve 15 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a) Dissolve 3 mg of tamoxifen citrate for performance test CRS (containing impurities A and F) in the mobile phase and dilute to 2.0 mL with the mobile phase.
Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Column:
- size: l = 0.25 m, Ø = 4.6 mm;
- stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 µm).
Mobile phase Mix 40 volumes of acetonitrile for chromatography R and 60 volumes of water for chromatography R containing 0.9 g/L of sodium dihydrogen phosphate R and 4.8 g/L of N,N-dimethyloctylamine R; adjust to pH 3.0 with phosphoric acid R.
Flow rate 1.2 mL/min.
Detection Spectrophotometer at 240 nm.
Injection 10 µL.
Run time Twice the retention time of tamoxifen.
Identification of impurities Use the chromatogram supplied with tamoxifen citrate for performance test CRS and the chromatogram obtained with reference solution (a) to identify the peaks due to impurities A and F.
Relative retention With reference to tamoxifen (retention time = about 20 min): impurity A = about 0.8; impurity F = about 0.9.
System suitability Reference solution (a):
- resolution: minimum 3.0 between the peaks due to impurities A and F; baseline separation between the peaks due to impurity F and tamoxifen.
Limits:
- impurity A: not more than 3 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.3 per cent);
- impurity F: not more than twice the area of the principal peak in the chromatogram obtained with reference solution (b) (0.2 per cent);
- unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (0.10 per cent);
- total: not more than 5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.5 per cent);
- disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.05 per cent); disregard any peak due to the citrate.
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on 1.000 g by drying in vacuo at 65 °C for 4 h.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.400 g in 75 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 56.36 mg of C32H37NO8.
IMPURITIES
Specified impurities A, F.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B, C, D, E, G, H.
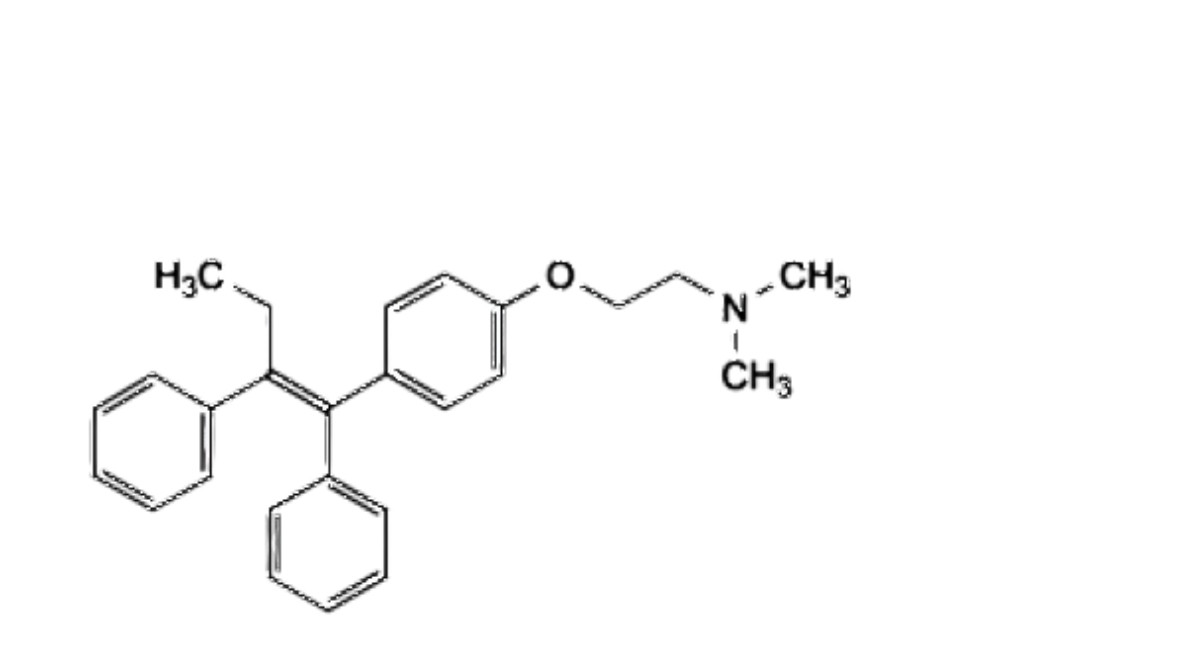
A. 2-[4-[(1 E)-1,2-diphenylbut-1-en-1-yl]phenoxy]-W,W-dimethylethan-1-amine ((E)-isomer),
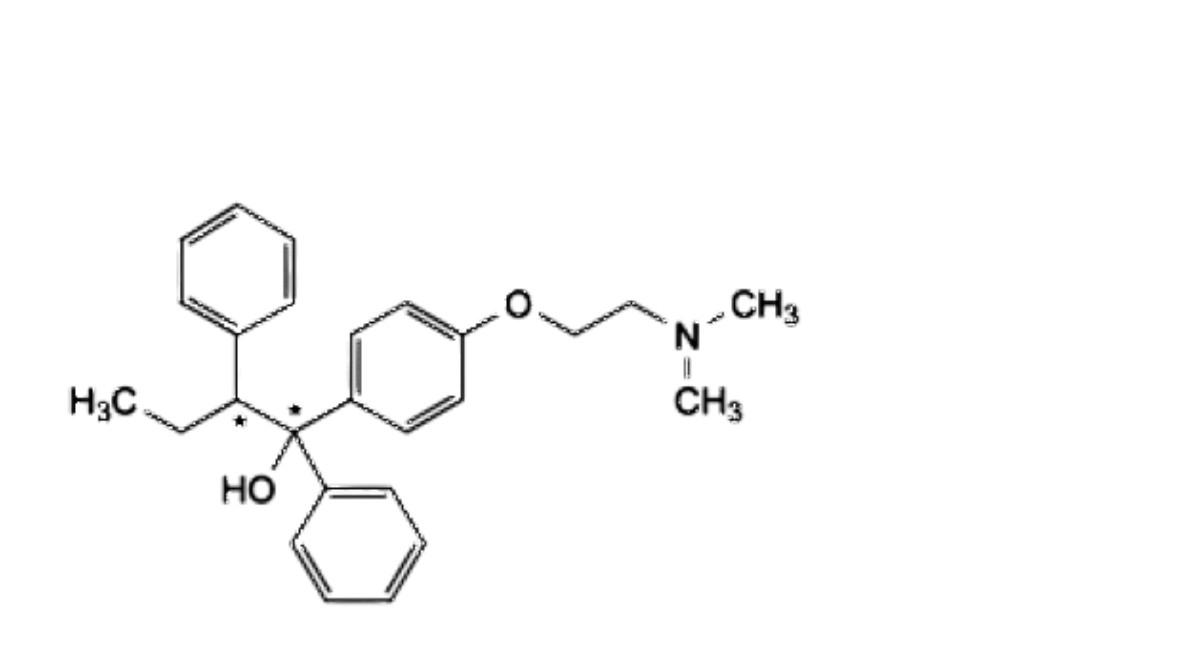
B.(1Ξ,2Ξ)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1,2-diphenylbutan-1-ol,
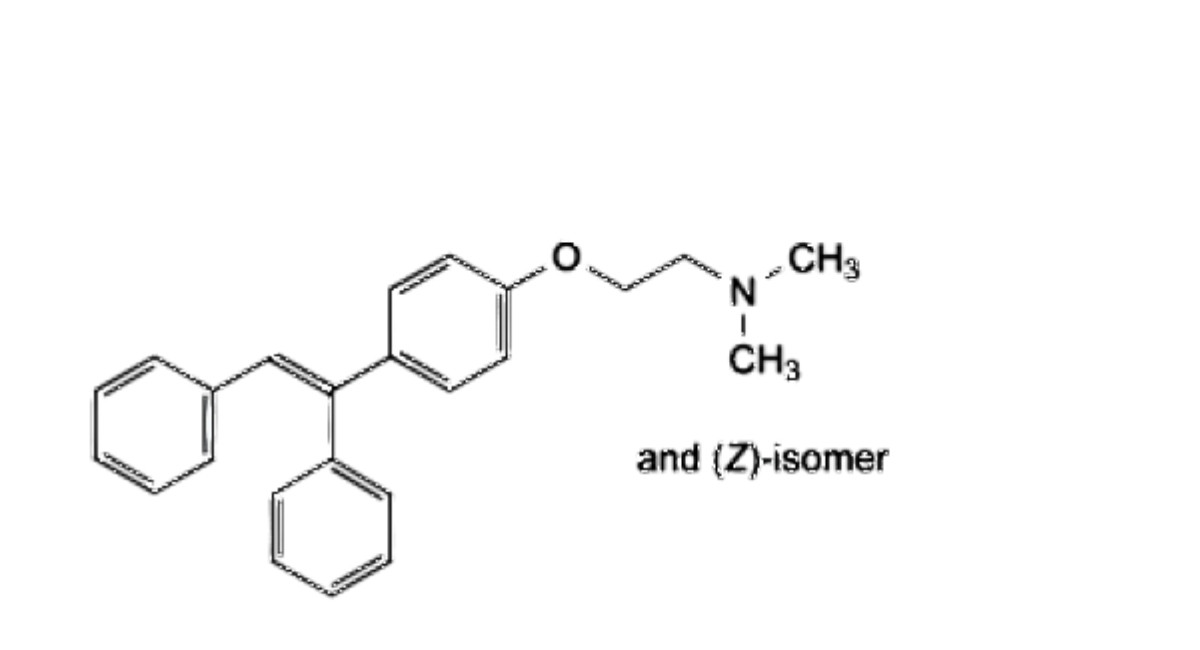
C. 2-[4-[(EZ)-1,2-diphenylethen-1-yl]phenoxy]-W,W-dimethylethan-1-amine,
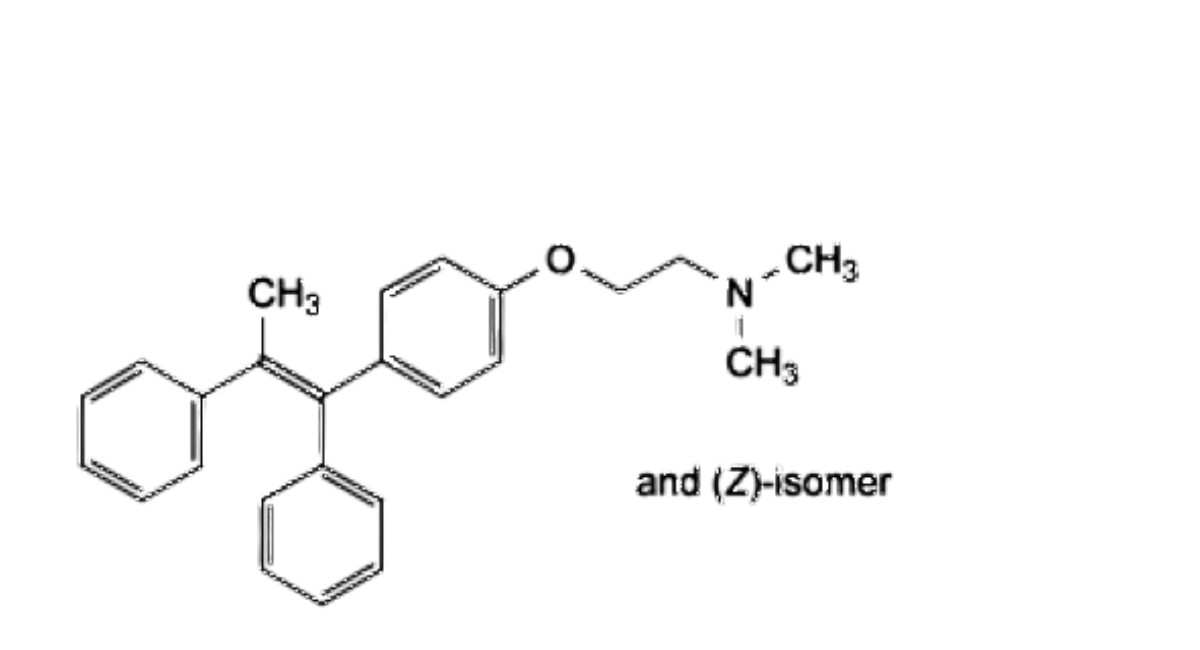
D. 2-[4-[(1 EZ)-1,2-diphenylprop-1-en-1-yl]phenoxy]-W,W-dimethylethan-1-amine,
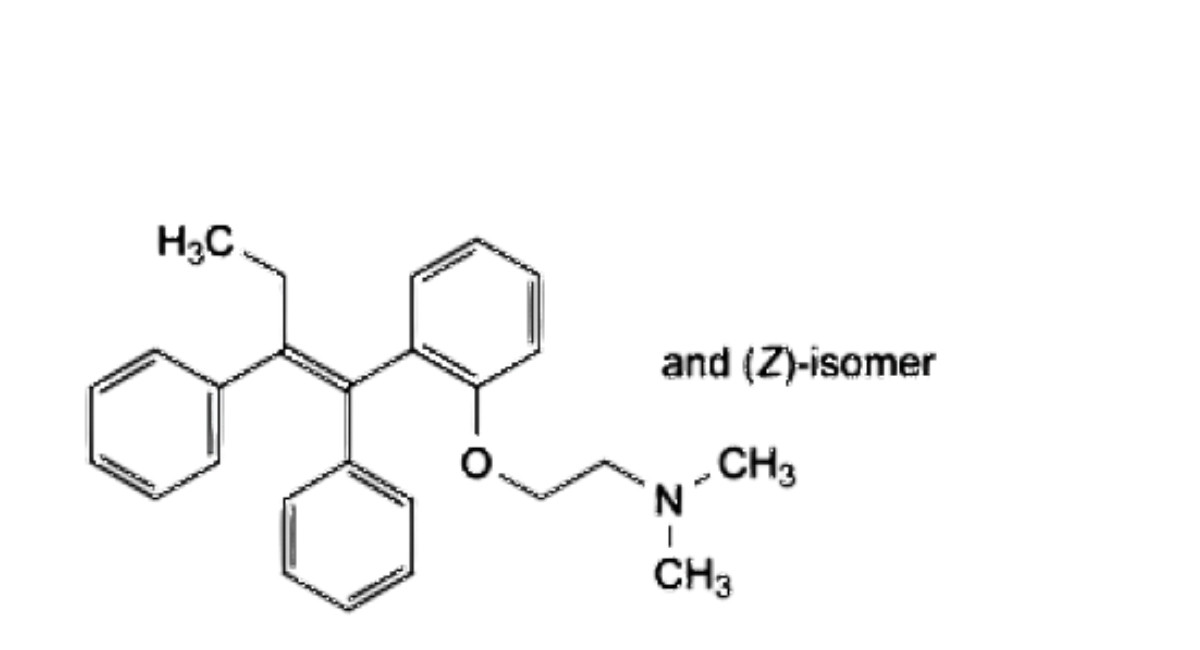
E. 2-[2-[(1 EZ)-1,2-diphenylbut-1-en-1-yl]phenoxy]-W,W-dimethylethan-1-amine,
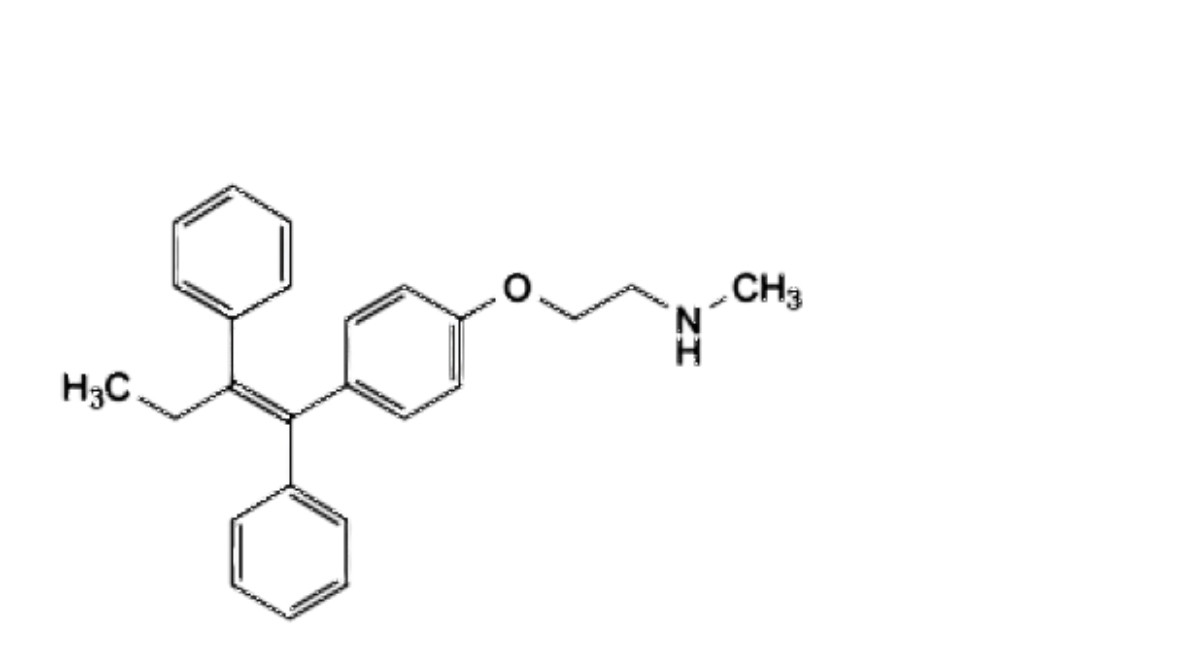
F. 2-[4-[(1 Z)-1,2-diphenylbut-1-en-1-yl]phenoxy]-W-methylethan-1-amine,
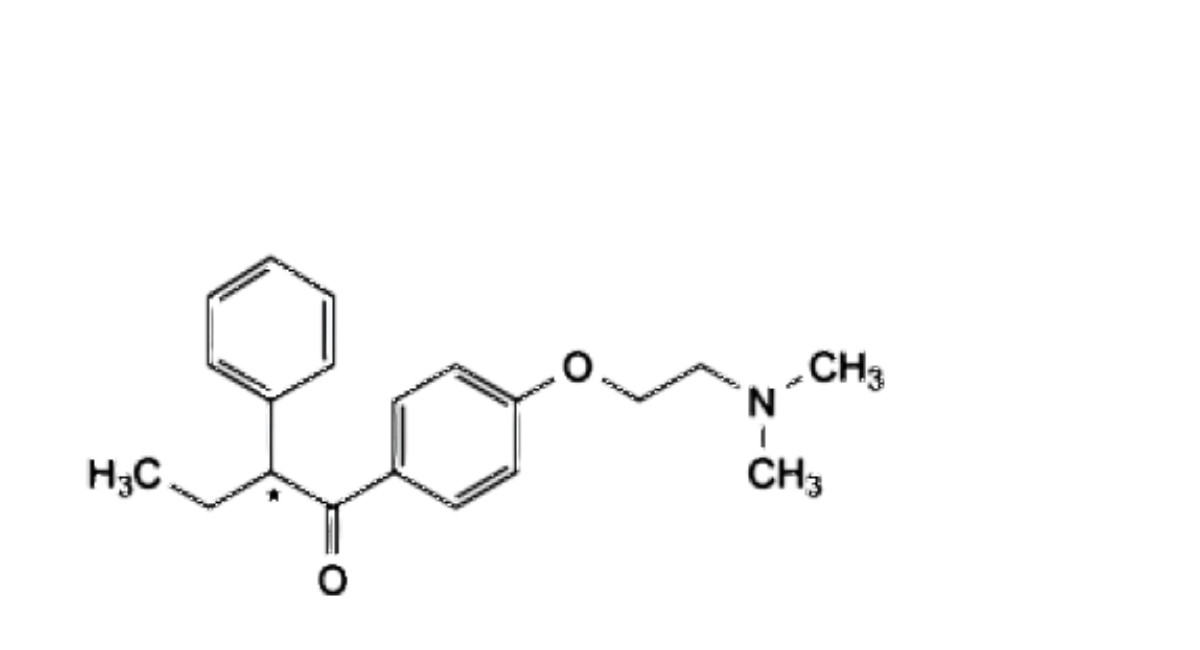
G. (2Ξ)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-2-phenylbutan-1-one,
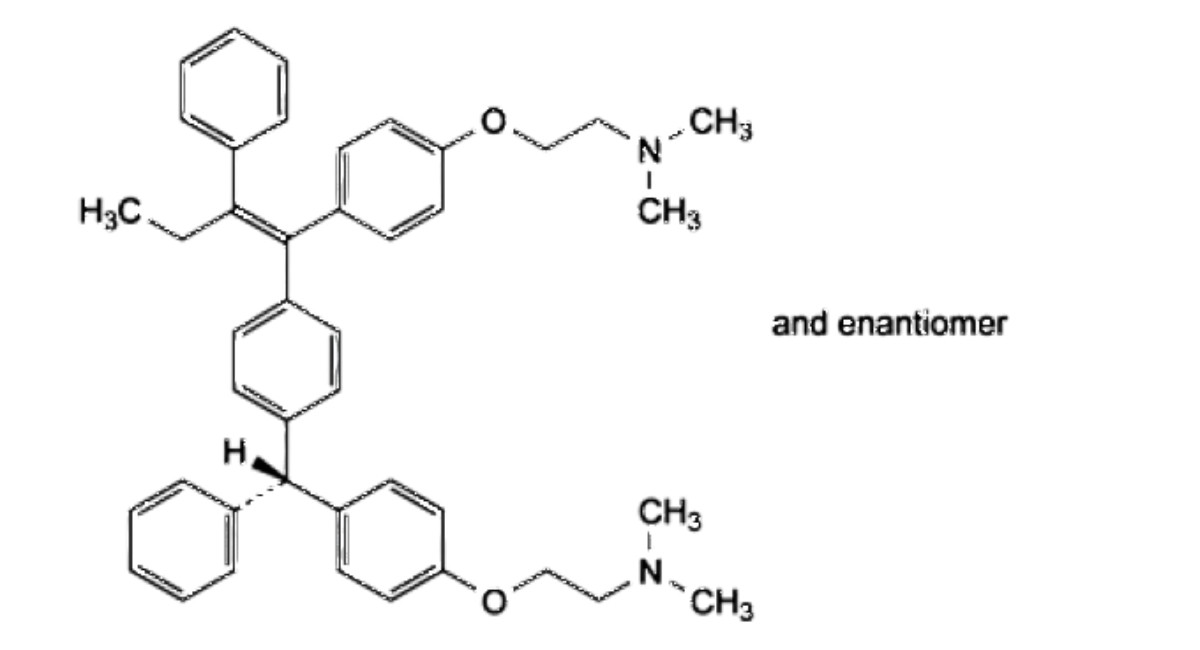
H. 2-[4-[(RS)-[4-[(1Z)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-2-phenylbut-1-en-1-yl]phenyl](phenyl)methyl]phenoxy]-W,W- dimethylethan-1-amine.
Ph Eur


