



Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Serotonin 5HT1 receptor agonist; treatment of migraine.
DEFINITION
Sumatriptan Nasal Spray is a solution of Sumatriptan containing suitable buffering agents in a suitable container fitted with a suitable nasal delivery system.
The nasal spray complies with the requirements stated under Nasal Preparations and with the following requirements.
Content of sumatriptan, C14H21N3O2S
90.0 to 110.0% of the stated amount.
CHARACTERISTICS
A clear, pale to dark yellow solution.
IDENTIFICATION
To a volume of nasal spray containing 40 mg of Sumatriptan add 1 mL of saturated sodium chloride solution and 1 mL of saturated sodium carbonate solution. Shake vigorously for 30 seconds, add two 2-mL quantities of propan-2-ol, shake, allow to separate and discard the aqueous layer. The separation may take up to 24 hours. Evaporate under a stream of nitrogen and dry at 100°.
The infrared absorption spectrum is concordant with the reference spectrum of sumatriptan (RS 414).
TESTS
Acidity
pH, 5.0 to 6.0, Appendix V L.
Impurities A and H
Carry out the method for liquid chromatography, Appendix III D. Prepare the solutions in a mixture containing 3 volumes of 0.025M sodium dihydrogen orthophosphate, the pH of which has been adjusted to 6.5 and 1 volume of acetonitrile.
(1) Dilute the nasal spray, if necessary, to contain 0.1% w/v of Sumatriptan.
(2) Dilute 1 volume of solution (1) to 200 volumes.
(3) Dilute the contents of a vial of sumatriptan for system suitability EPCRS to 1 mL.
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (25 cm × 4.6 mm) packed with silica gel for chromatography (5 µm) (Spherisorb silica S5W is suitable).
(b) Use isocratic elution and the mobile phase described below.
(c) Use a flow rate of 2.0 mL per minute.
(d) Use an ambient column temperature.
(e) Use a detection wavelength of 282 nm.
(f) Inject 20 µL of each solution.
(g) Allow the chromatography to proceed for 5 times the retention time of the principal peak.
MOBILE PHASE
10 volumes of 10M ammonium acetate and 90 volumes of methanol.
SYSTEM SUITABILITY
The test is not valid unless the chromatogram obtained with solution (3) resembles that supplied with sumatriptan for system suitability EPCRS and the resolution between impurity A and sumatriptan is at least 1.5.
LIMITS
Identify any peak in the chromatogram obtained with solution (1) due to impurity A using the chromatogram obtained with solution (3). Multiply the area of any peak corresponding to impurity A by a correction factor of 0.6.
In the chromatogram obtained with solution (1):
the area of any peak corresponding to impurity A is not greater than 3 times the area of the principal peak obtained in the chromatogram obtained with solution (2) (1.5%);
the area of any peak corresponding to impurity H is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (0.5%).
Related substances
Carry out the method for liquid chromatography, Appendix III D. Prepare the solutions in a mixture containing 3 volumes of 0.025M sodium dihydrogen orthophosphate, the pH of which has been adjusted to 6.5, and 1 volume of acetonitrile.
(1) Dilute the nasal spray, if necessary, to contain 0.1% w/v of Sumatriptan.
(2) Dilute 1 volume of solution (1) to 100 volumes.
(3) Dilute the contents of a vial of sumatriptan impurity mixture EPCRS to 1 mL.
(4) Dilute 1 volume of solution (2) to 10 volumes.
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (25 cm × 4.6 mm) packed with octadecylsilyl silica gel for chromatography (5 µm) (Spherisorb ODS 1 is suitable).
(b) Use isocratic elution and the mobile phase below.
(c) Use a flow rate of 1.5 mL per minute.
(d) Use an ambient column temperature.
(e) Use a detection wavelength of 282 nm.
(f) Inject 20 µL of each solution.
(g) For solution (1) allow the chromatography to proceed for 4 times the retention time of the principal peak.
MOBILE PHASE
25 volumes of acetonitrile and 75 volumes of a solution containing 0.97 g of dibutylamine, 0.735 g of orthophosphoric acid and 2.93 g of sodium dihydrogen orthophosphate in 750 mL water, adjusted to pH to 6.5 with 10M sodium hydroxide and diluted to 1000 mL with water.
When the chromatograms are recorded under the prescribed conditions the retention times relative to sumatriptan (retention time about 17 minutes) are: impurity D, about 0.2; impurity 2, about 0.3; impurity 1, about 0.4; impurity E, about 0.5; impurity B, about 0.6; impurity F, about 0.7 and impurity C, about 0.8.
SYSTEM SUITABILITY
The test is not valid unless, in the chromatogram obtained with solution (3), the resolution between the peaks due to impurity C and sumatriptan is at least 1.5.
LIMITS
Identify any peaks in the chromatogram obtained with solution (1) due to impurity 1 and impurity 2 using the reference chromatogram for sumatriptan impurity standard BPCRS.
Multiply the area of any peak corresponding to impurity 1 by a correction factor of 0.2 and multiply the area of any peak corresponding to impurity 2 by a correction factor of 0.3.
In the chromatogram obtained with solution (1):
the area of any peak corresponding to impurity 1 is not greater than 1.5 times the area of the principal peak in the chromatogram obtained with solution (2) (1.5%);
the area of any peak corresponding to impurity 2 is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (1.0%);
the area of any other secondary peak is not greater than half of the area of the principal peak in the chromatogram obtained with solution (2) (0.5%).
Disregard any peak with an area less than the area of the principal peak in the chromatogram obtained with solution (4) (0.1%).
The total impurity content in the test for Impurities A and H and the test for Related substances is not greater than 4.0%.
ASSAY
Carry out the method for liquid chromatography, Appendix III D. Prepare the solutions in a mixture containing 3 volumes of 0.025M sodium dihydrogen orthophosphate, the pH of which has been adjusted to 6.5, and 1 volume of acetonitrile.
(1) Dilute the nasal spray, if necessary, to contain 0.05% w/v of Sumatriptan.
(2) 0.07% w/v of sumatriptan succinate BPCRS.
(3) Dilute the contents of a vial of sumatriptan impurity mixture EPCRS to 1 mL.
CHROMATOGRAPHIC CONDITIONS
The chromatographic procedure described under Related substances may be used.
SYSTEM SUITABILITY
The test is not valid unless, in the chromatogram obtained with solution (3), the resolution between the peaks due to impurity C and sumatriptan is at least 1.5.
DETERMINATION OF CONTENT
Calculate the content of C14H21N3O2S using the declared content of C14H21N3O2S,C4H6O4 in sumatriptan succinate BPCRS. 1 mg of C14H21N3O2S,C4H6O4 is equivalent to 0.714 mg of C14H21N3O2S.
IMPURITIES
The impurities limited by the requirements of this monograph include those listed under Sumatriptan Succinate and the following:
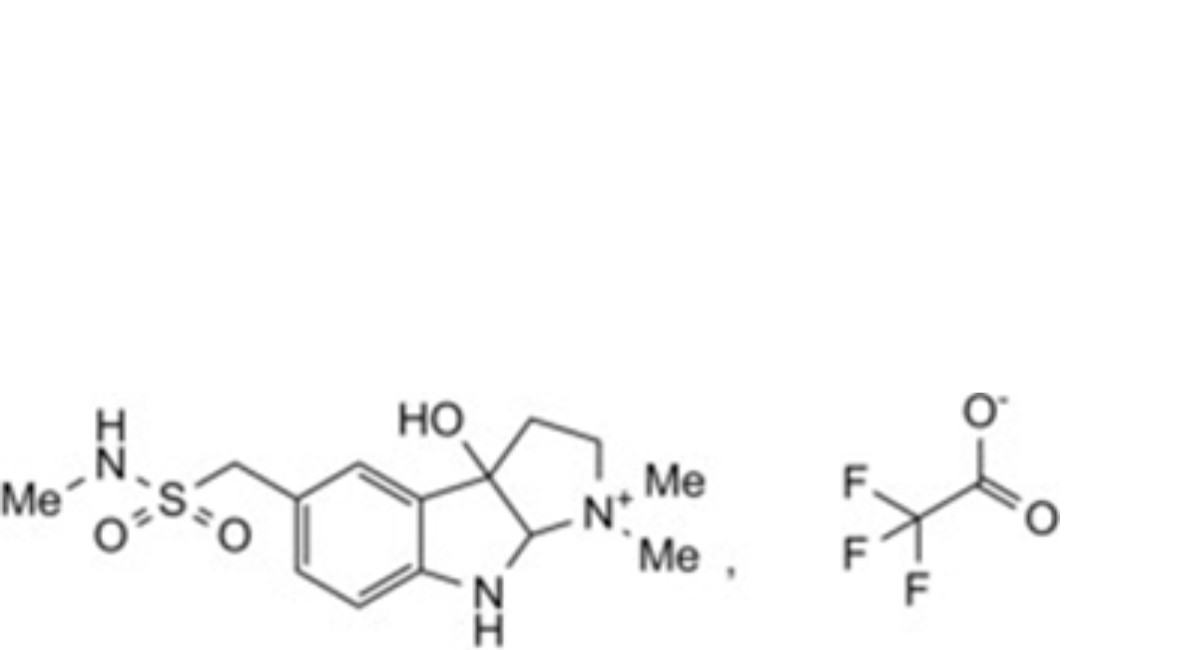
1. 3a-hydroxy-1,1-dimethyl-5-{[methylamino)sulfonyl]methyl}-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-1-ium trifluoroacetate];
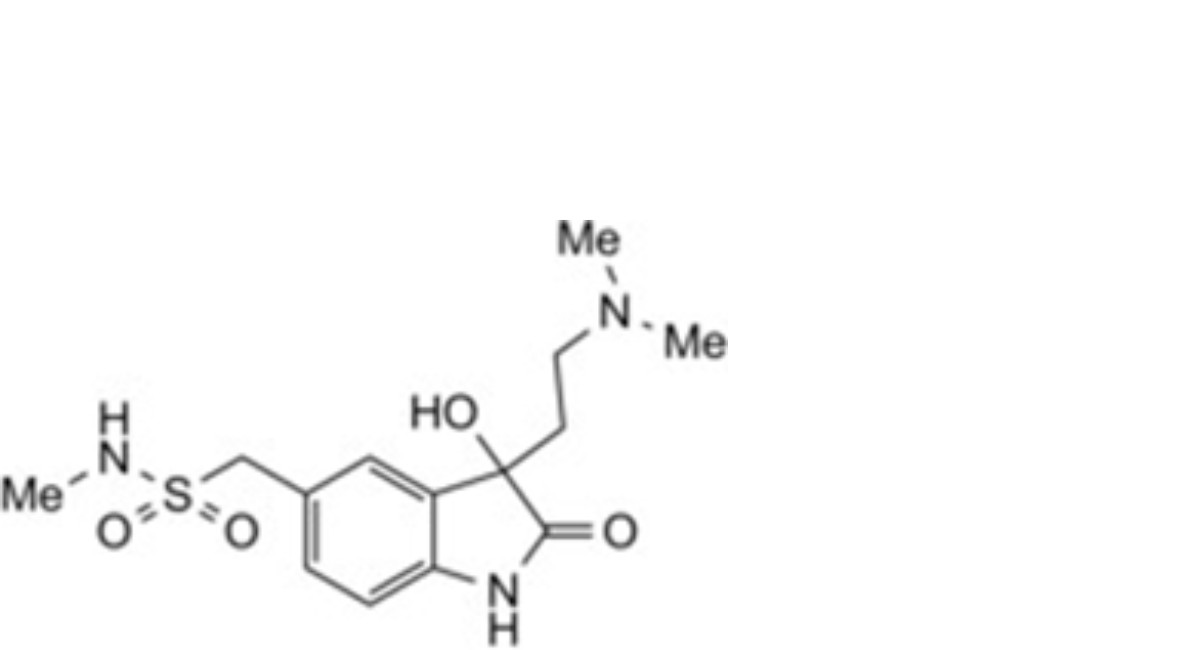
2. 1-{3-[2-(dimethylnitroyl)ethyl]-1H-indol-5-yl}-N-methylmethanesulfonamide.


