


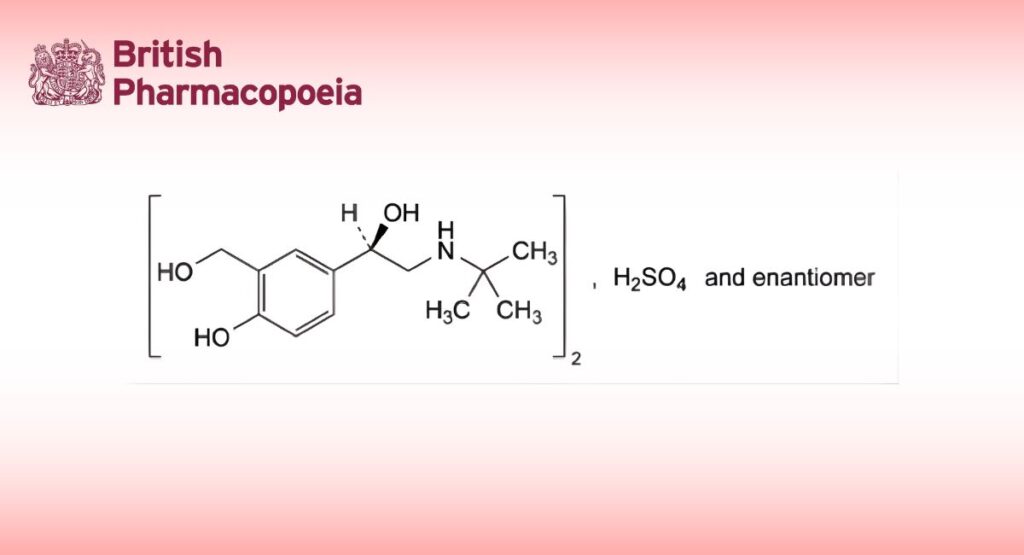
Salbutamol Sulphate
(Ph. Eur. monograph 0687)
C26H44N2O10S 576.7 51022-70-9
Action and use
Beta2-adrenoceptor agonist; bronchodilator.
Preparations
Salbutamol Inhalation Powder
Salbutamol Inhalation Powder, pre-metered
Salbutamol Injection
Salbutamol Nebuliser Solutionư
Salbutamol Oral Solutionư
Salbutamol Pressurised Inhalationư
Salbutamol Tablets
DEFINITION
Bis[4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol] sulfate.
Content
98.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Freely soluble in water, practically insoluble or very slightly soluble in ethanol (96 per cent) and in methylene chloride.
IDENTIFICATION
First identification: A, C.
Second identification: B, C.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison: salbutamol sulfate CRS.
B. Thin-layer chromatography (2.2.27).
Test solution: Dissolve 12 mg of the substance to be examined in water R and dilute to 10 mL with the same solvent.
Reference solution: Dissolve 12 mg of salbutamol sulfate CRS in water R and dilute to 10 mL with the same solvent.
Plate: TLC silica gel plate R.
Mobile phase: concentrated ammonia R, water R, ethyl acetate R, 2-propanol R, methyl isobutyl ketone R (3:18:35:45:50 V/V/V/V/V).
Application: 1 μL.
Development: Over 2/3 of the plate.
Drying: In air.
Detection: Spray with a 1 g/L solution of methylbenzothiazolone hydrazone hydrochloride R in a 90 per cent V/V solution of methanol R, followed by a 20 g/L solution of potassium ferricyanide R in a mixture of 1 volume of concentrated ammonia R1 and 3 volumes of water R, followed by a further spraying with a 1 g/L solution of methylbenzothiazolone hydrazone hydrochloride R in a 90 per cent V/V solution of methanol R.
Results: The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with the reference solution.
C. It gives reaction (a) of sulfates (2.3.1).
TESTS
Solution S
Dissolve 0.25 g in carbon dioxide-free water R and dilute to 25 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and not more intensely coloured than reference solution BY6 (2.2.2, Method II).
Acidity or alkalinity
To 10 mL of solution S add 0.15 mL of methyl red solution R and 0.2 mL of 0.01 M sodium hydroxide. The solution is yellow. Not more than 0.4 mL of 0.01 M hydrochloric acid is required to change the colour of the indicator to red.
Related substances
Liquid chromatography (2.2.29). Protect the solutions from light.
Test solution: Dissolve 25.0 mg of the substance to be examined in mobile phase A and dilute to 50.0 mL with mobile phase A.
Reference solution (a): Dilute 1.0 mL of the test solution to 100.0 mL with mobile phase A. Dilute 1.0 mL of this solution to 10.0 mL with mobile phase A.
Reference solution (b): Dissolve 2 mg of salbutamol for peak identification CRS (containing impurities C, F, N, O and P) in 5 mL of mobile phase A.
Reference solution (c): With the aid of ultrasound, dissolve the contents of a vial of salbutamol impurity J CRS in 1 mL of the test solution.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: end-capped solid core octylsilyl silica gel for chromatography R (2.7 μm);
— temperature: 30 °C.
Mobile phase:
— mobile phase A: dissolve 3.45 g of sodium dihydrogen phosphate monohydrate R in about 900 mL of water for chromatography R and add 0.5 mL of triethylamine R; adjust to pH 3.0 with dilute phosphoric acid R and dilute to 1000 mL with water for chromatography R;
— mobile phase B: methanol R, acetonitrile R (35:65 V/V);
| Time (min) |
Mobile phase A (per cent V/V) |
Mobile phase B (per cent V/V) |
| 0 – 5 | 95 | 5 |
| 5 – 25 | 95 → 70 | 5 → 30 |
| 25 – 27 | 70 | 30 |
| 27 – 30 | 70 → 10 | 30 → 90 |
| 30 – 35 | 10 | 90 |
Flow rate 1.0 mL/min.
Detection Spectrophotometer at 273 nm.
Injection 20 μL.
Identification of impurities Use the chromatogram supplied with salbutamol for peak identification CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities C, F, N, O and P; use the
chromatogram obtained with reference solution (c) to identify the peak due to impurity J.
Relative retention With reference to salbutamol (retention time = about 5 min): impurity J = about 0.92; impurity C = about 2.3; impurity N (isomer 1) = about 2.84; impurity N (isomer 2) = about 2.86; impurity P = about 2.91; impurity F = about 3.03; impurity O = about 3.07.
System suitability:
— resolution: minimum 1.5 between the peaks due to impurities F and O in the chromatogram obtained with reference solution (b);
— peak-to-valley ratio: minimum 2.0, where Hp = height above the baseline of the peak due to impurity J and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to salbutamol in the chromatogram obtained with reference solution (c); minimum 2.0, where Hp = height above the baseline of the peak due to impurity N (isomer 1) and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to impurity N (isomer 2) in the chromatogram obtained with reference solution (b); minimum 2.0, where Hp = height above the baseline of the peak due to impurity N (isomer 2) and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to impurity P in the chromatogram obtained with reference solution (b).
Calculation of percentage contents:
— for each impurity, use the concentration of salbutamol sulfate in reference solution (a).
Limits:
— impurity F: maximum 0.3 per cent;
— impurities C, O: for each impurity, maximum 0.15 per cent;
— impurity N: for the sum of the areas of the peaks due to the 2 isomers, maximum 0.15 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 1.0 per cent;
— reporting threshold: 0.05 per cent.
Boron
Maximum 50 ppm.
Test solution To 50 mg of the substance to be examined add 5 mL of a solution containing 13 g/L of anhydrous sodium carbonate R and 17 g/L of potassium carbonate R. Evaporate to dryness on a water-bath and dry at 120 °C. Ignite the residue rapidly until the organic matter has been destroyed, allow to cool and add 0.5 mL of water R and 3.0 mL of a freshly prepared 1.25 g/L solution of curcumin R in glacial acetic acid R. Warm gently to effect solution, allow to cool and add 3.0 mL of a mixture prepared by adding 5 mL of sulfuric acid R, slowly and with stirring, to 5 mL of glacial acetic acid R. Mix and allow to stand for 30 min. Dilute to 100.0 mL with ethanol (96 per cent) R, filter and use the filtrate.
Reference solution Dissolve 0.572 g of boric acid R in 1000.0 mL of water R. Dilute 1.0 mL of the solution to 100.0 mL with water R. To 2.5 mL of this solution add 5 mL of a solution containing 13 g/L of anhydrous sodium carbonate R and 17 g/L of potassium carbonate R, and treat this mixture in the same manner as the test solution.
Measure the absorbance (2.2.25) of the test solution and of the reference solution at the absorption maximum at about 555 nm. The absorbance of the test solution is not greater than that of the reference solution.
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.400 g in 5 mL of formic acid R and add 35 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 57.67 mg of C26H44N2O10S.
STORAGE
Protected from light.
IMPURITIES
Specified impurities C, F, N, O.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) A, B, D, E, G, H, I, J, K, L, M, P, Q.
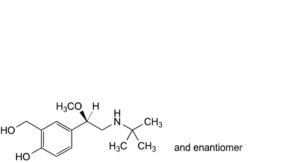
A. 4-[(1RS)-2-(tert-butylamino)-1-methoxyethyl]-2-(hydroxymethyl)phenol,
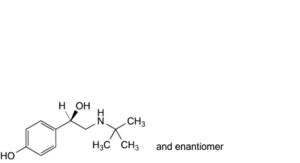
B. 4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]phenol,
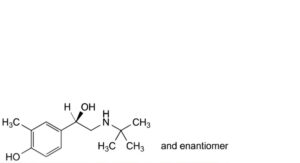
C. 4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-methylphenol,
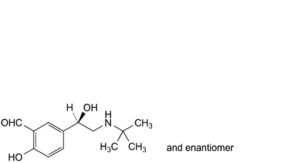
D. 5-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-hydroxybenzaldehyde,
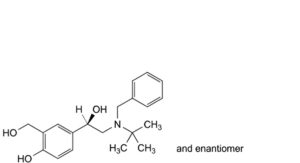
E. 4-[(1RS)-2-[benzyl(tert-butyl)amino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol,
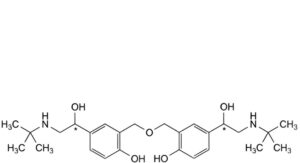
F. 2,2′-[oxybis(methylene)]bis[4-[(1Ξ)-2-(tert-butylamino)-1-hydroxyethyl]phenol],
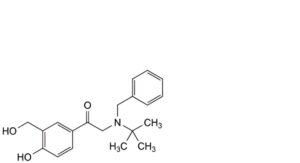
G. 2-[benzyl(tert-butyl)amino]-1-[4-hydroxy-3-(hydroxymethyl)phenyl]ethan-1-one,
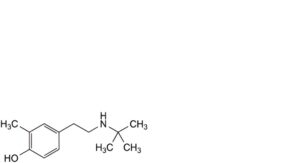
H. 4-[2-(tert-butylamino)ethyl]-2-methylphenol,
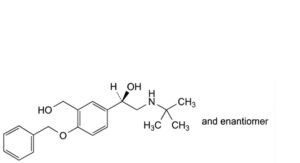
I. (1RS)-1-[4-(benzyloxy)-3-(hydroxymethyl)phenyl]-2-(tert-butylamino)ethan-1-ol,
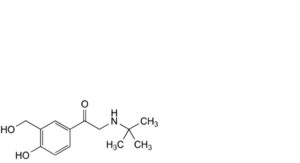
J. 2-(tert-butylamino)-1-[4-hydroxy-3-(hydroxymethyl)phenyl]ethan-1-one (salbutamone),
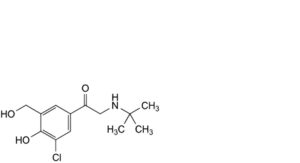
K. 2-(tert-butylamino)-1-[3-chloro-4-hydroxy-5-(hydroxymethyl)phenyl]ethan-1-one,
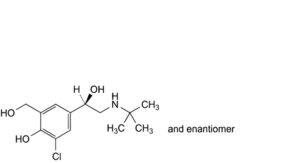
L. 4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-chloro-6-(hydroxymethyl)phenol,
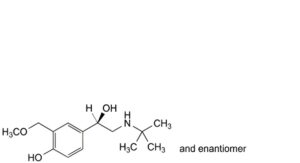
M. 4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-(methoxymethyl)phenol,
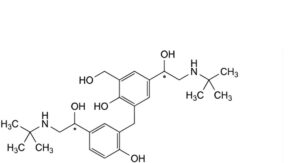
N. 4-[(1Ξ)-2-(tert-butylamino)-1-hydroxyethyl]-2-[[5-[(1Ξ)-2-(tert-butylamino)-1-hydroxyethyl]-2-hydroxyphenyl]methyl]-6-(hydroxymethyl)phenol,
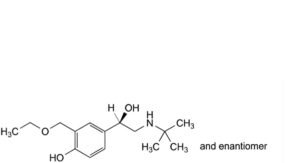
O. 4-[(1RS)-2-(tert-butylamino)-1-hydroxyethyl]-2-(ethoxymethyl)phenol,
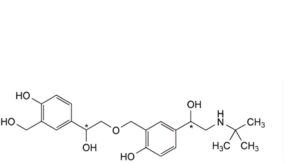
P. 4-[(1Ξ)-2-(tert-butylamino)-1-hydroxyethyl]-2-[[(2Ξ)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethoxy]methyl]phenol,
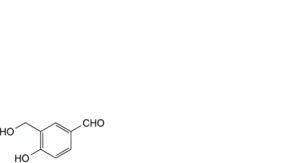
Q. 4-hydroxy-3-(hydroxymethyl)benzaldehyde.


