



Action and use
Beta2-adrenoceptor agonist; bronchodilator.
DEFINITION
Salbutamol Pressurised Inhalation, Suspension is a suspension of Salbutamol Sulfate in a suitable liquid in a pressurised container fitted with a metering dose valve.
The pressurised inhalation, suspension complies with the requirements stated under Preparations for Inhalation and with the following requirements.
PRODUCTION
The size of aerosol particles to be inhaled is controlled so that a consistent portion is deposited in the lungs. The fine-particle characteristics of preparations for inhalation are determined using the method described in Appendix XII C7.
Preparations for inhalation: Aerodynamic Assessment of Fine Particles. The test and limits should be agreed with the competent authority.
The water content is controlled to ensure the performance of the product as justified and authorised by the competent authority.
Content of salbutamol, C13H21NO3
85.0 to 115.0% of the stated delivered dose (ex-actuator).
IDENTIFICATION
Discharge the inhaler a sufficient number of times into a mortar to obtain the equivalent of 2 mg of salbutamol, grind the residue thoroughly with 0.1 g of potassium bromide, add a further 0.2 g of potassium bromide and mix thoroughly. The infrared absorption spectrum, Appendix II A, is concordant with the reference spectrum of salbutamol sulfate (RS 315).
TESTS
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions prepared in mobile phase A.
(1) Shake a quantity of the pressurised inhalation containing the equivalent of 1 mg of salbutamol with 10 mL of mobile phase A, mix with the aid of ultrasound and filter.
(2) Dilute 1 volume of solution (1) to 20 volumes and further dilute 1 volume to 10 volumes.
(3) 0.012% w/v of salbutamol sulfate BPCRS, 0.00005% w/v of salbutamol ketone BPCRS (impurity J) and 0.00002% w/v of salbutamol impurity Q BPCRS.
(4) 0.012% w/v of salbutamol for peak identification EPCRS.
(5) 0.012% w/v of salbutamol impurity standard BPCRS.
(6) Dilute 1 volume of solution (2) to 5 volumes.
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (15 cm × 4.6 mm) packed with base-deactivated end-capped octylsilyl silica gel for chromatography (3 μm) (Hypersil BDS C8 is suitable).
(b) Use gradient elution and the mobile phase described below.
(c) Use a flow rate of 1.0 mL per minute.
(d) Use a column temperature of 30°.
(e) Use a detection wavelength of 273 nm.
(f) Inject 50 μL of each solution.
MOBILE PHASE
Mobile phase A 0.5 volumes of triethylamine and 1000 volumes of 0.025M sodium dihydrogen orthophosphate, adjust to pH 3.0 with 10% v/v of orthophosphoric acid.
Mobile phase B 350 volumes of methanol and 650 volumes of acetonitrile.
| Time (Minutes) | Mobile phase A (% v/v) | Mobile phase B (% v/v) | Comment |
| 0-5 | 95 | 5 | isocratic |
| 5-18 | 95→70 | 5→30 | linear gradient |
| 18-20 | 70 | 30 | isocratic |
| 20-20.1 | 70→10 | 30→90 | linear gradient |
| 20.1-25 | 10 | 90 | isocratic |
| 25-25.1 | 10→95 | 90→5 | linear gradient |
| 25.1-33 | 95 | 5 | re-equilibration |
SYSTEM SUITABILITY
The test is not valid unless: in the chromatogram obtained with solution (3), the peak-to-valley ratio is at least 6.0, where Hp is the height above the baseline of the peak due to impurity J and Hv is the height above the baseline of the lowest point of the curve separating this peak from the peak due to salbutamol;
in the chromatogram obtained with solution (6), the signal-to-noise ratio for the peak due to salbutamol is at least 20.
CALCULATION OF IMPURITIES
For impurities J and Q, use the concentration of each impurity in solution (3).
For all other impurities, use the concentration of salbutamol in solution (2).
For the reporting threshold, use the concentration of salbutamol in solution (6). For impurity N, apply the reporting threshold to the sum of impurity N peaks 1 and 2.
For peak identification, use solutions (3), (4) and (5).
Salbutamol retention time: about 8 minutes.
Relative retention: impurity J, about 0.95; impurity Q, about 1.4; impurity D, about 1.7; impurity N (peak 1), about 1.77; impurity N (peak 2), about 1.79; impurity F, about 1.9.
Correction factors: impurity D, multiply by 1.5.
LIMITS
— impurities D and F: for each impurity, not more than 0.5%;
— impurities N (sum of peaks 1 and 2) and Q: for each impurity, not more than 0.2%;
— unspecified impurities: for each impurity, not more than 0.2%;
— total impurities: not more than 1.5%;
— reporting threshold: 0.1%.
Uniformity of delivered dose
Complies with the requirements stated under Pressurised Metered-dose Preparations for Inhalation using the following method of analysis. Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
(1) Collect single doses of the preparation being examined using the procedure described under Pressurised Metered-dose Preparations for Inhalation, Uniformity of delivered dose and dissolve the collected dose in sufficient methanol to produce a solution containing the equivalent of 0.000075% w/v of salbutamol.
(2) 0.000092% w/v of salbutamol sulfate BPCRS in methanol.
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (10 cm × 3 mm) packed with end-capped octadecylsilyl silica gel for chromatography (5 μm) (Spherisorb ODS is suitable).
(b) Use isocratic elution and the mobile phase described below.
(c) Use a flow rate of 0.85 mL per minute.
(d) Use a column temperature of 50°.
(e) Use a detection wavelength of 276 nm.
(f) Inject 150 μL of each solution.
MOBILE PHASE
238 volumes of 0.1% w/v of ammonium acetate solution and 762 volumes of methanol.
When the chromatograms are recorded under the prescribed conditions, the retention time of salbutamol is about 2 minutes.
SYSTEM SUITABILITY
The test is not valid, unless in the chromatogram obtained with solution (2), the symmetry factor for the peak due to salbutamol is between 0.8 to 2.5.
DETERMINATION OF CONTENT
Calculate the content of salbutamol, C13H21NO3, per delivered dose using the declared content of C13H21NO3 in salbutamol sulfate BPCRS. Repeat the procedure as described under Pressurised Metered-dose Preparations for Inhalation, Uniformity of delivered dose.
ASSAY
Use the average of the individual results determined in the test for Uniformity of delivered dose.
LABELLING
The quantity of active ingredient is stated in terms of the equivalent amount of salbutamol.
IMPURITIES
The impurities limited by the requirements of this monograph include impurity C, D, F, J, K, M, N, O and Q listed under Salbutamol Sulfate and:
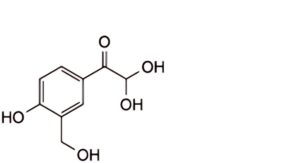
1. 2,2-dihydroxy-1-[4-hydroxy-3-(hydroxymethyl)phenyl]ethanone (glyoxal impurity)
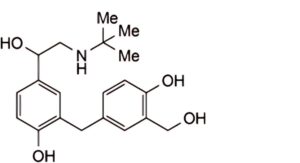
2. 2-[4-hydroxy-3-(hydroxymethyl)benzyl]-4-{1-hydroxy-2-[(2-methyl-2-propanyl)amino]ethyl}phenol (head-to-tail dimer impurity).


