



(Ph Eur. monograph 3008)
Action and use
HMG Co-A reductase inhibitor; lipid-regulating drug.
DEFINITION
Tablets containing Rosuvastatin calcium (2631), for human use.
They comply with the monograph Tablets (0478) and the following additional requirements.
Content
93.0 per cent to 105.0 per cent of the content of rosuvastatin (C22H28FN3O6S) stated on the label.
IDENTIFICATION
A. Record the UV spectrum of the principal peak in the chromatograms obtained with the solutions used in the assay, with a diode array detector in the range of 210-400 nm. Results
The UV spectrum of the principal peak in the chromatogram obtained with the test solution is similar to the UV spectrum of the principal peak in the chromatogram obtained with reference solution (a).
B. Examine the chromatograms obtained in the assay.
Results: The principal peak in the chromatogram obtained with the test solution is similar in retention time and size to the principal peak in the chromatogram obtained with reference solution (a).
TESTS
Related substances
Liquid chromatography (2.2.29). Carry out the test protected from light and prepare the solutions immediately before use.
Solvent mixture acetonitrile R, water R (25:75 V/V).
Test solution: To an appropriate number of tablets (at least 6) add at least 50 mL of water R and shake vigorously until the tablets have disintegrated completely. Add a suitable volume of acetonitrile R and again shake vigorously. Add a suitable volume of water R to obtain a ratio of acetonitrile to water of 25:75 V/V. Filter through a poly(vinylidene difluoride) filter and dilute with the solvent mixture, if necessary, to obtain a concentration of rosuvastatin calcium of 1 mg/mL.
Reference solution (a): To 25.0 mg of rosuvastatin calcium CRS add 25 mL of water R and allow to stand for at least 10 min. Sonicate for about 30 min until dissolved, shaking every 10 min. Add 12.5 mL of acetonitrile R, mix well and dilute to 50.0 mL with water R. Dilute 1.0 mL of the solution to 50.0 mL with the solvent mixture.
Reference solution (b): Dissolve 7 mg of rosuvastatin for system suitability CRS (containing impurities A, B and C) in 2.5 mL of acetonitrile R and dilute to 10 mL with water R.
Reference solution (c): Dissolve the contents of a vial of rosuvastatin impurity mixture CRS (containing impurity D) in 1 mL of the solvent mixture.
Reference solution (d): Dissolve 2 mg of rosuvastatin ethyl ester R (impurity FP-A) in 20 mL of the solvent mixture. Dilute 1 mL of the solution to 100 mL with the solvent mixture.
Column:
— size: l = 0.15 m, Ø = 3.0 mm;
— stationary phase: end-capped octadecylsilyl amorphous organosilica polymer for chromatography R (3.5 μm);
— temperature: 40 °C.
Mobile phase:
— mobile phase A: 1 per cent V/V solution of trifluoroacetic acid R, acetonitrile for chromatography R, water for chromatography R (1:31:68 V/V/V);
— mobile phase B: 1 per cent V/V solution of trifluoroacetic acid R, water for chromatography R, acetonitrile for chromatography R (1:37:66 V/V/V);
| Time
(min) |
Mobile phase A
(per cent V/V) |
Mobile phase B
(per cent V/V) |
| 0 – 26 | 100 | 0 |
| 26 – 36 | 100 → 20 | 0 → 80 |
| 36 – 46 | 20 | 80 |
Flow rate: 0.7 mL/min.
Detection: Spectrophotometer at 242 nm.
Injection: 10 μL.
Identification of impurities: Use the chromatogram supplied with rosuvastatin for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities A, B and C; use the chromatogram supplied with rosuvastatin impurity mixture CRS and the chromatogram obtained with reference solution (c) to identify the peak due to impurity D; use the chromatogram obtained with reference solution (d) to identify impurity FP-A.
Relative retention: With reference to rosuvastatin (retention time = about 11 min): impurity A = about 0.9; impurity B = about 1.1; impurity C = about 1.7; impurity D = about 2.2; impurity FP-A = about 3.1.
System suitability: Reference solution (b):
— peak-to-valley ratio: minimum 2.0, where Hp = height above the baseline of the peak due to impurity B and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to rosuvastatin.
Calculation of percentage contents:
— correction factor: multiply the peak area of impurity C by 1.4;
— for each impurity, use the concentration of rosuvastatin calcium in reference solution (a).
Limits:
— impurities C, D: for each impurity, maximum 1.5 per cent;
— impurity FP-A: maximum 0.5 per cent;
— unspecified impurities: for each impurity, maximum 0.2 per cent;
— total: maximum 2.5 per cent;
— reporting threshold: 0.1 per cent; disregard the peaks due to impurities A and B.
Dissolution1 (2.9.3, Apparatus 2)
Carry out the test protected from light.
Dissolution medium (0.05 M citrate buffer solution pH 6.6): Dissolve 147.0 g of sodium citrate R in 2 L of water R, add 3.3 g of anhydrous citric acid R and dilute to 10 L with the same solvent. If necessary, adjust to pH 6.6 ± 0.05 with sodium citrate R or anhydrous citric acid R. Use 900 mL of the medium.
Rotation speed: 50 r/min.
Time: 30 min.
Analysis Liquid chromatography (2.2.29).
Test solution: The samples withdrawn from the dissolution vessel and filtered, using a poly(vinylidene difluoride) filter.
Reference solution: To 25.0 mg of rosuvastatin calcium CRS add 25 mL of water R and allow to stand for at least 10 min.
Sonicate for about 30 min until dissolved, shaking every 10 min. Add 12.5 mL of acetonitrile R, mix well and dilute to 50.0 mL with water R. Dilute a suitable volume of the solution with the dissolution medium to obtain a concentration of rosuvastatin corresponding to the theoretical concentration of rosuvastatin in the test solution, based on the labelled content of the tablets.
Column:
— size: l = 0.05 m, Ø = 4.6 mm;
— stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase phosphoric acid R, acetonitrile for chromatography R, water for chromatography R (0.1:40:60 V/V/V).
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 242 nm.
Injection: 20 μL.
Run time: 5 min.
System suitability: Reference solution:
— repeatability: maximum relative standard deviation of 1.0 per cent determined on 6 injections.
Calculate the amount of dissolved rosuvastatin (C22H28FN3O6S), expressed as a percentage of the content stated on the label, taking into account the assigned content of rosuvastatin calcium CRS and applying a conversion factor of 0.96.
Acceptance criterion:
— Q = 75 per cent after 30 min.
ASSAY
Liquid chromatography (2.2.29). Carry out the test protected from light.
Solvent mixture acetonitrile R, water R (25:75 V/V).
Test solution: To an appropriate number of tablets (at least 6) add at least 50 mL of water R and shake vigorously until the tablets have disintegrated completely. Add a suitable volume of acetonitrile R and again shake vigorously. Add a suitable volume of water R to obtain a ratio of acetonitrile to water of 25:75 V/V. Filter through a poly(vinylidene difluoride) filter and dilute with the solvent mixture, if necessary, to obtain a concentration of rosuvastatin calcium of 0.1 mg/mL.
Reference solution (a): To 25.0 mg of rosuvastatin calcium CRS add 25 mL of water R and allow to stand for at least 10 min. Sonicate for about 30 min until dissolved, shaking every 10 min. Add 12.5 mL of acetonitrile R, mix well and dilute to 50.0 mL with water R. Dilute 1.0 mL of the solution to 5.0 mL with the solvent mixture.
Reference solution (b): Dissolve 7 mg of rosuvastatin for system suitability CRS (containing impurities A, B and C) in 2.5 mL of acetonitrile R and dilute to 10 mL with water R.
Column:
— size: l = 0.15 m, Ø = 3.0 mm;
— stationary phase: end-capped octadecylsilyl amorphous organosilica polymer for chromatography R (3.5 μm);
— temperature: 40 °C.
Mobile phase:
— mobile phase A: 1 per cent V/V solution of trifluoroacetic acid R, acetonitrile for chromatography R, water for chromatography R (1:31:68 V/V/V);
— mobile phase B: 1 per cent V/V solution of trifluoroacetic acid R, acetonitrile for chromatography R (1:100 V/V);
| Time
(min) |
Mobile phase A
(per cent V/V) |
Mobile phase B
(per cent V/V) |
| 0 – 14 | 100 | 0 |
| 14 – 15 | 100 → 10 | 0 → 90 |
Flow rate: 0.7 mL/min.
Detection: Spectrophotometer at 242 nm.
Injection: 10 μL.
Identification of impurities: Use the chromatogram supplied with rosuvastatin for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peak due to impurity B.
Relative retention: With reference to rosuvastatin (retention time = about 11 min): impurity B = about 1.1.
System suitability:
— repeatability: maximum relative standard deviation of 1.0 per cent determined on 6 injections of reference solution (a);
— peak-to-valley ratio: minimum 2.0, where Hp = height above the baseline of the peak due to impurity B and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to rosuvastatin in the chromatogram obtained with reference solution (b). Calculate the percentage content of rosuvastatin (C22H28FN3O6S) taking into account the assigned content of rosuvastatin calcium CRS and applying a conversion factor of 0.96.
IMPURITIES
Specified impurities C, D, FP-A.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph): A, B, FP-B.
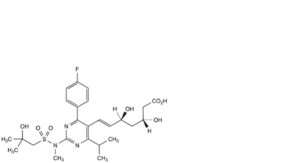
A. (3R,5S,6E)-7-[2-(2,N-dimethyl-2-hydroxypropane-1-sulfonamido)-4-(4-fluorophenyl)-6-(propan-2-yl)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoic acid,
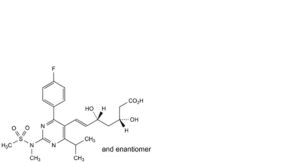
B. (3RS,5RS,6E)-7-[4-(4-fluorophenyl)-2-(N-methylmethanesulfonamido)-6-(propan-2-yl)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoic acid,
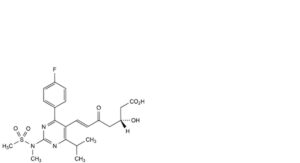
C. (3R,6E)-7-[4-(4-fluorophenyl)-2-(N-methylmethanesulfonamido)-6-(propan-2-yl)pyrimidin-5-yl]-3-hydroxy-5-oxohept-6-enoic acid,
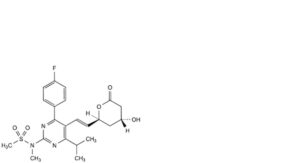
D. N-[4-(4-fluorophenyl)-5-[(1E)-2-[(2S,4R)-4-hydroxy-6-oxooxan-2-yl]ethen-1-yl]-6-(propan-2-yl)pyrimidin-2-yl]-N-methylmethanesulfonamide,
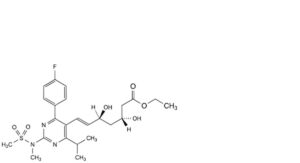
FP-A. ethyl (3R,5S,6E)-7-[4-(4-fluorophenyl)-2-(N-methylmethanesulfonamido)-6-(propan-2-yl)pyrimidin-5-yl]-3,5-dihydroxyhept-6-enoate (rosuvastatin ethyl ester),
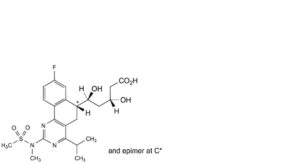
FP-B. (3R,5S)-5-[(6RS)-8-fluoro-2-(N-methylmethanesulfonamido)-4-(propan-2-yl)-5,6-dihydrobenzo[h]quinazolin-6-yl]-3,5-dihydroxypentanoic acid.
1The test approved in the marketing authorisation is to be used for routine quality control to confirm batch-to-batch consistency. For more
information please consult Ph. Eur. 1. General Notices.


