



Effervescent Ranitidine Tablets
Action and use
Histamine H2 receptor antagonist; treatment of peptic ulcer disease.
DEFINITION
Ranitidine Effervescent Tablets contain Ranitidine Hydrochloride in a suitable effervescent basis.
The tablets comply with the requirements stated under Tablets and with the following requirements.
Content of ranitidine, C13H22N4O3S
95.0 to 105.0% of the stated amount.
IDENTIFICATION
A. The light absorption, Appendix II B, in the range 200 to 400 nm of the solution prepared by dissolving one tablet in 100 mL of water exhibits a maximum at 313 nm. Dilute the solution if necessary.
B. In the Assay, the retention time of the principal peak in the chromatogram obtained with solution (1) is the same as that of the principal peak in the chromatogram obtained with solution (2).
C. Dissolves with vigorous effervescence on the addition of warm water to produce a clear solution.
TESTS
Disintegration
Comply with the requirement for Effervescent Tablets stated under Tablets.
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
(1) Shake a quantity of powdered tablets containing the equivalent of 0.13 g of Ranitidine with 100 mL methanol (10%) and filter (Whatman No. 42. paper is suitable).
(2) Dilute 1 volume of solution (1) to 500 volumes with methanol (10%).
(3) 0.0065% w/v of ranitidine for impurity A identification EPCRS in mobile phase A.
(4) Dissolve the contents of a vial of ranitidine impurity J EPCRS in 1 mL of solution (1).
(5) Dilute 1 volume of solution (2) to 2 volumes with methanol (10%).
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (10 cm × 4.6 mm) packed with octadecylsilyl amorphous organosilica polymer (3.5 μm) (Waters Xterra MS C18 is suitable).
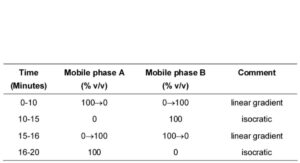
(b) Use gradient elution and the mobile phase described below.
(c) Use a flow rate of 1.5 mL per minute.
(d) Use a column temperature of 35°.
(e) Use a detection wavelength of 230 nm.
(f) Inject 20 μL of each solution.
MOBILE PHASE
Buffer solution Adjust the pH of 950 mL of 0.05M potassium dihydrogen orthophosphate to 7.1 by adding strong sodium hydroxide solution and dilute to 1 litre.
Mobile phase A: 2 volumes of acetonitrile and 98 volumes of the buffer solution.
Mobile phase B: 22 volumes of acetonitrile and 78 volumes of the buffer solution.
SYSTEM SUITABILITY
The test is not valid unless, in the chromatogram obtained with solution (4), the resolution between the two principal peaks is at least 1.5.
LIMITS
Identify any peak due to impurity A in the chromatogram obtained with solution (1), using the chromatogram obtained with solution (3).
In the chromatogram obtained with solution (1):
the area of any secondary peak with a retention time relative to ranitidine of 0.66 is not greater than 2.5 times the area of the principal peak in the chromatogram obtained with solution (2) (0.5%);
the area of any peak corresponding to impurity A is not greater than 2.5 times the area of the principal peak in the chromatogram obtained with solution (2) (0.5%);
the area of any other secondary peak is not greater than the area of the principal peak in the chromatogram obtained with solution (2) (0.2%);
the total area of any such peaks is not greater than 5 times the area of the principal peak in the chromatogram obtained with solution (2) (1.0%).
Disregard any peak with an area less than that of the principal peak in the chromatogram obtained with solution (5) (0.1%).
ASSAY
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
(1) Shake 10 tablets with 400 mL of methanol (10%) until the tablets have completely disintegrated, dilute to 500 mL with methanol (10%), filter (Whatman GF/C paper is suitable) and dilute the filtrate with the mobile phase to produce a solution containing the equivalent of 0.01% w/v of ranitidine.
(2) 0.0112% w/v of ranitidine hydrochloride BPCRS in methanol (10%).
(3) 0.0112% w/v of ranitidine hydrochloride BPCRS and 0.0002% w/v of dimethyl{5-[2-(1-methylamino-2-nitrovinylamino)ethylsulfinylmethyl]furfuryl}amine BPCRS in methanol (10%)
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (25 cm × 4.6 mm) packed with octadecylsilyl silica gel for chromatography (10 μm) (Partisil ODS 1 is suitable).
(b) Use isocratic elution and the mobile phase described below.
(c) Use a flow rate of 2 mL per minute.
(d) Use ambient column temperature.
(e) Use a detection wavelength of 322 nm.
(f) Inject 20 μL of each solution.
MOBILE PHASE
15 volumes of 0.1M ammonium acetate and 85 volumes of methanol.
SYSTEM SUITABILITY
The assay is not valid unless the peak due to ranitidine in the chromatogram obtained with solution (3) shows baseline separation from the peak due to dimethyl{5-[2-(1-methylamino-2-nitrovinylamino)ethylsulfinylmethyl]-furfuryl}amine.
DETERMINATION OF CONTENT
Calculate the content of C13H22N4O3S in the tablets using the declared content of C13H22N4O3S in ranitidine hydrochloride BPCRS.
STORAGE
Ranitidine Effervescent Tablets should be protected from light.
LABELLING
The quantity of active ingredient is stated in terms of the equivalent amount of ranitidine.
IMPURITIES
The impurities limited by the requirements of this monograph include those listed under Ranitidine Hydrochloride.


