


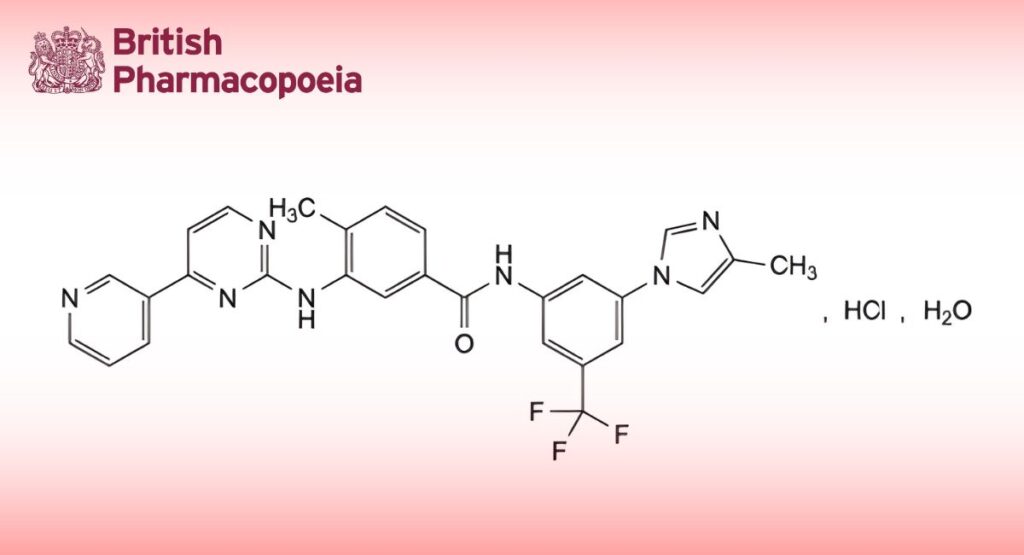
(Ph. Eur. monograph 2993)
C28H23ClF3N7O,H2O 584.0 923288-90-8
Action and use
Tyrosine kinase (BCR-ABL) inhibitor; treatment of chronic myeloid leukaemia.
DEFINITION
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzamide hydrochloride monohydrate.
Content
98.0 per cent to 102.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or slightly yellowish or slightly greenish-yellow, hygroscopic, crystalline powder.
Solubility
Practically insoluble in water, slightly soluble in anhydrous ethanol, very slightly soluble in heptane.
It shows polymorphism (5.9).
IDENTIFICATION
A. Infrared absorption spectrophotometry (2.2.24).
Comparison: nilotinib hydrochloride monohydrate CRS.
If the spectra obtained in the solid state show differences, dissolve the substance to be examined and the reference substance separately in anhydrous ethanol R, evaporate to dryness and record new spectra using the residues.
B. It gives reaction (b) of chlorides (2.3.1).\
C. Water (see Tests).
TESTS
Impurity A
Liquid chromatography (2.2.29). Carry out the test protected from light.
Solvent mixture: dimethyl sulfoxide R, water R (20:80 V/V).
Test solution: Dissolve 0.300 g of the substance to be examined in 2 mL of dimethyl sulfoxide R, add 7 mL of water R, allow to equilibrate at room temperature without shaking to avoid foam formation, and then dilute to 10.0 mL with water R. Shake well, allow the substance to be examined to precipitate for about 2 h in the dark and filter the supernatant through a 0.45 μm filter.
Reference solution (a): Dissolve 7.5 mg of nilotinib impurity A CRS in dimethyl sulfoxide R and dilute to 100.0 mL with the same solvent. Dilute 1.0 mL of the solution to 100.0 mL with dimethyl sulfoxide R. Dilute 2.0 mL of this solution to 10.0 mL with water R.
Reference solution (b): Dissolve 7.5 mg of nilotinib impurity B CRS and 7.5 mg of nilotinib impurity C CRS in dimethyl sulfoxide R and dilute to 100.0 mL with the same solvent. Dilute 1.0 mL of the solution to 100.0 mL with dimethyl sulfoxide R. Dilute 2.0 mL of this solution to 10.0 mL with water R.
Reference solution (c): Dilute 2.0 mL of reference solution (b) to 10.0 mL with the solvent mixture.
Column:
— size: l = 0.15 m, Ø = 3.0 mm;
— stationary phase: encapsulated polar-embedded octadecylsilyl silica gel for chromatography R (3 μm);
— temperature: 40 °C.
Mobile phase:
— mobile phase A: 1.36 g/L solution of potassium dihydrogen phosphate R adjusted to pH 3.0 with phosphoric acid R;
— mobile phase B: mobile phase A, acetonitrile R1 (20:80 V/V);
| Time (min) |
Mobile phase A (per cent V/V) |
Mobile phase B (per cent V/V) |
| 0 – 2 | 85 | 15 |
| 2 – 10 | 85 → 80 | 15 → 20 |
| 10 – 12 | 80 → 75 | 20 → 25 |
| 12 – 18 | 75 → 10 | 25 → 90 |
| 18 – 19 | 10 | 90 |
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 207 nm.
Injection: 20 μL of the test solution and reference solution (a).
Identification of impurities: Use the chromatogram obtained with reference solution (a) to identify the peak due to impurity A.
Relative retention: With reference to nilotinib (retention time = about 17 min): impurity A = about 0.3.
System suitability:
— repeatability: maximum relative standard deviation of 5.0 per cent determined on 6 injections of reference solution (a).
Calculation of content:
— for impurity A, use the concentration of impurity A in reference solution (a).
Limit:
— impurity A: maximum 3 ppm.
Impurities B and C
Liquid chromatography (2.2.29) as described in the test for impurity A with the following modifications.
Detection: Spectrophotometer at 225 nm.
Injection: Test solution and reference solutions (b) and (c).
Identification of impurities: Use the chromatogram obtained with reference solution (b) to identify the peaks due to impurities B and C.
Relative retention: With reference to nilotinib (retention time = about 17 min): impurity C = about 0.2; impurity B = about 0.6.
System suitability:
— signal-to-noise ratio: minimum 10 for the peaks due to impurities B and C in the chromatogram obtained with reference solution (c);
— repeatability: maximum relative standard deviation of 5.0 per cent for the areas of the peaks due to impurities B and C, determined on 6 injections of reference solution (b).
Calculation of contents:
— for impurities B and C, use the concentration of the corresponding impurity in reference solution (b).
Limits:
— impurities B, C: for each impurity, maximum 2 ppm.
Related substances
Liquid chromatography (2.2.29). Carry out the test protected from light.
Solvent mixture: anhydrous ethanol R, water R (50:50 V/V).
Test solution: Dissolve 20.0 mg of the substance to be examined in the solvent mixture and dilute to 200.0 mL with the solvent mixture.
Reference solution (a): Dissolve 20.0 mg of nilotinib hydrochloride monohydrate CRS in the solvent mixture and dilute to 200.0 mL with the solvent mixture.
Reference solution (b): Dilute 1.0 mL of the test solution to 100.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 10.0 mL with the solvent mixture.
Reference solution (c): Dissolve 2 mg of nilotinib for system suitability CRS (containing impurities E, F and G) in the solvent mixture and dilute to 20 mL with the solvent mixture.
Column:
— size: l = 0.15 m, Ø = 3.0 mm;
— stationary phase: end-capped octadecylsilyl silica gel for chromatography compatible with 100 per cent aqueous mobile phases R (3 μm);
— temperature: 40 °C.
Mobile phase:
— mobile phase A: 1.36 g/L solution of potassium dihydrogen phosphate R adjusted to pH 3.0 with phosphoric acid R;
— mobile phase B: mobile phase A, acetonitrile for chromatography R (20:80 V/V);
| Time (min) |
Mobile phase A (per cent V/V) |
Mobile phase B (per cent V/V) |
| 0 – 2 | 90 | 10 |
| 2 – 16 | 90 → 10 | 10 → 90 |
| 16 – 17 | 10 | 90 |
Flow rate: 0.8 mL/min.
Detection: Spectrophotometer at 240 nm.
Injection: 5 μL of the test solution and reference solutions (b) and (c).
Identification of impurities: Use the chromatogram supplied with nilotinib for system suitability CRS and the chromatogram obtained with reference solution (c) to identify the peaks due to impurities E, F and G.
Relative retention: With reference to nilotinib (retention time = about 12 min): impurity E = about 1.03; impurity F = about 1.07; impurity G = about 1.09.
System suitability: Reference solution (c):
— resolution: minimum 1.5 between the peaks due to impurities F and G;
— peak-to-valley ratio: minimum 2.0, where Hp = height above the baseline of the peak due to impurity E and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to nilotinib.
Calculation of percentage contents:
— for each impurity, use the concentration of nilotinib hydrochloride monohydrate in reference solution (b).
Limits:
— impurity F: maximum 0.2 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 0.40 per cent;
— reporting threshold: 0.05 per cent.
Water (2.5.12)
3.0 per cent to 5.0 per cent, determined on 0.150 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g in a platinum crucible.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modification.
Injection Test solution and reference solution (a).
Calculate the percentage content of C28H23ClF3N7O,H2O taking into account the assigned content of nilotinib hydrochloride monohydrate CRS.
STORAGE
In an airtight container.
IMPURITIES
Specified impurities A, B, C, F.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) D, E, G, H.
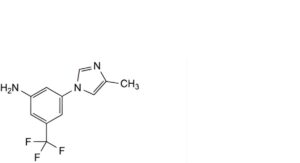
A. 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline,
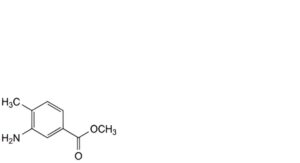
B. methyl 3-amino-4-methylbenzoate,
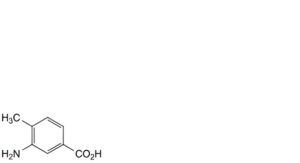
C. 3-amino-4-methylbenzoic acid,
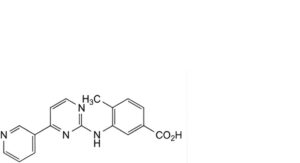
D. 4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzoic acid,
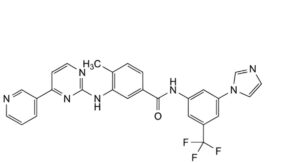
E. N-[3-(1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzamide,
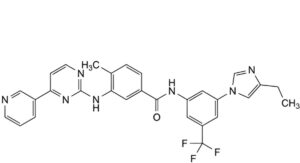
F. N-[3-(4-ethyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzamide,
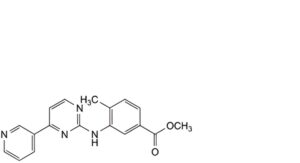
G. methyl 4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzoate,
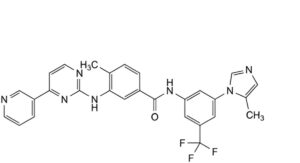
H. 4-methyl-N-[3-(5-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]benzamide.


