


Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Cholinesterase inhibitor.
Preparation
Neostigmine Injection Ph Eur
DEFINITION
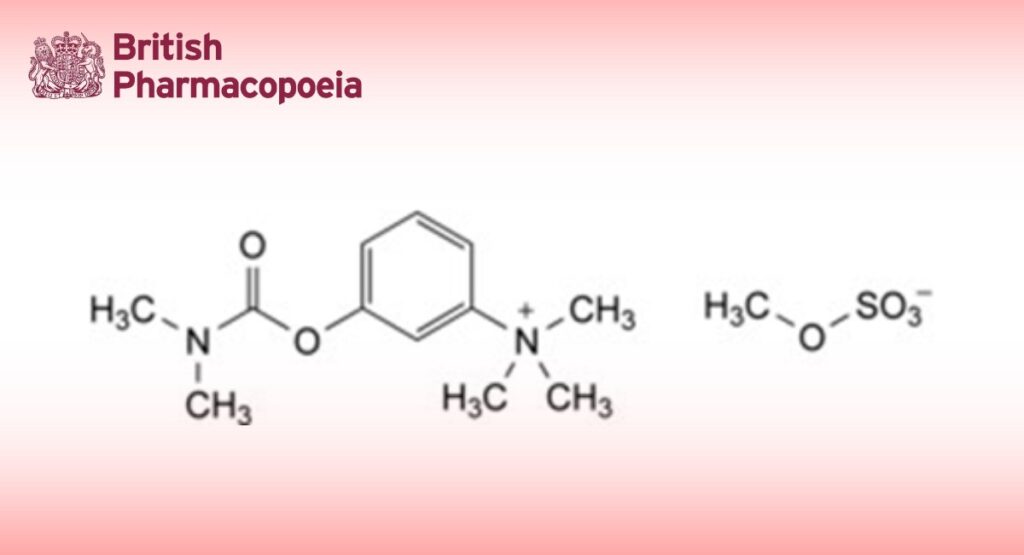
3-[(Dimethylcarbamoyl)oxy]-N,N,N-trimethylanilinium methyl sulfate.
Content
98.5 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder or colourless crystals, hygroscopic.
Solubility
Very soluble in water, freely soluble in ethanol (96 per cent).
IDENTIFICATION
First identification: A, C.
Second identification: A, B, D, E.
A. Melting point (2.2.14): 144 °C to 149 °C.
B. Ultraviolet and visible absorption spectrophotometry (2.2.25).
Test solution Dissolve 50 mg in 0.5 M sulfuric acid and dilute to 100 mL with the same acid.
Spectral range 230-350 nm.
Absorption maxima 261 nm and 267 nm.
Resolution (2.2.25): minimum 1.9 for the absorbance ratio.
Absorbance ratio A267/A261 = 0.84 to 0.87.
C. Infrared absorption spectrophotometry (2.2.24).
Comparison neostigmine metilsulfate CRS.
D. To 50 mg add 0.4 g of potassium hydroxide R and 2 mL of ethanol (96 per cent) R and heat on a water- bath for 3 min, replacing the evaporated ethanol (96 per cent). Cool and add 2 mL of water R and 2 mL of diazobenzenesulfonic acid solution R1. An orange-red colour develops.
E. Dissolve 0.1 g in 5 mL of distilled water R and add 1 mL of barium chloride solution R1. No precipitate is formed. Add 2 mL of hydrochloric acid R and heat in a water-bath for 10 min. A fine, white precipitate is formed.
TESTS
Solution S
Dissolve 2.5 g in distilled water R and dilute to 50 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and colourless (2.2.2, Method II).
Acidity or alkalinity
To 4.0 mL of solution S add 6.0 mL of water R and 0.1 mL of phenolphthalein solution R1. The solution is colourless. Add 0.3 mL of 0.01 M sodium hydroxide; the solution becomes red. Add 0.4 mL of 0.01 M hydrochloric acid; the solution becomes colourless. Add 0.1 mL of methyl red solution R; the solution becomes red or yellowish-red.
Related substances
Liquid chromatography (2.2.29).
Test solution Dissolve 50.0 mg of the substance to be examined in the mobile phase and dilute to 50.0 mL with the mobile phase.
Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b) Dissolve 4 mg of 3-dimethylaminophenol R (impurity B) in 50 mL of the mobile phase. Dilute 1 mL of the solution to 200 mL with the mobile phase.
Reference solution (c) Dissolve the contents of a vial of neostigmine impurity A CRS in 1 mL of reference solution (b).
Reference solution (d) Mix 1 mL of the mobile phase and 1 mL of reference solution (a).
Column:
— size: l = 0.25 m, Ø = 4.0 mm;
— stationary phase: base-deactivated octylsilyl silica gel for chromatography R (5 µm);
— temperature: 30 °C.
Mobile phase To 710 mL of a 3.6 g/L solution of sodium dihydrogen phosphate R previously adjusted to pH 3.2 with phosphoric acid R, add 4.3 g of sodium dodecyl sulfate R and 290 mL of acetonitrile for chromatography R.
Flow rate 1.6 mL/min.
Detection Spectrophotometer at 220 nm.
Injection 50 µL of the test solution and reference solutions (a), (c) and (d).
Run time Twice the retention time of neostigmine.
Identification of impurities Use the chromatogram obtained with reference solution (c) to identify the peaks due to impurities A and B.
Relative retention With reference to neostigmine (retention time = about 20 min): impurity B = about 0.56; impurity A = about 0.61.
System suitability:
— resolution: minimum 1.5 between the peaks due to impurities B and A in the chromatogram obtained with reference solution (c);
— signal-to-noise ratio: minimum 25 for the principal peak in the chromatogram obtained with reference solution (d).
Calculation of percentage contents:
— correction factor: multiply the peak area of impurity B by 0.5;
— for each impurity, use the concentration of neostigmine metilsulfate in reference solution (a).
Limits:
— impurity B: maximum 0.01 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 0.2 per cent;
— reporting threshold: 0.05 per cent, except for impurity B.
Sulfates (2.4.13)
Maximum 200 ppm, determined on solution S.
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.300 g in 150 mL of water R and add 100 mL of dilute sodium hydroxide solution R. Distil, collecting the distillate in 40 mL of a 40 g/L solution of boric acid R until the total volume in the collecting vessel is about 250 mL. Titrate the solution in the collecting vessel with 0.1 M hydrochloric acid, using
0.25 mL of methyl red mixed solution R as indicator. Carry out a blank test. 1 mL of 0.1 M hydrochloric acid is equivalent to 33.44 mg of C13H22N2O6S.
STORAGE
In an airtight container, protected from light.
IMPURITIES
Specified impurities B.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10.
Control of impurities in substances for pharmaceutical use) A, C.
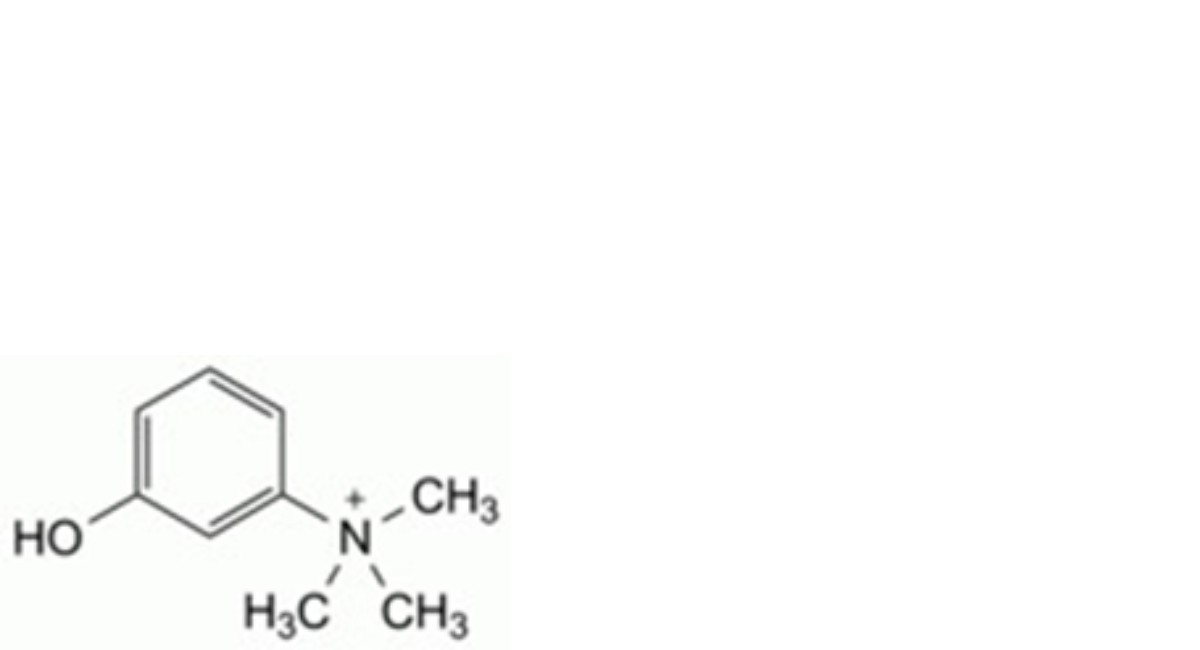
A. 3-hydroxy-N,N,N-trimethylanilinium,
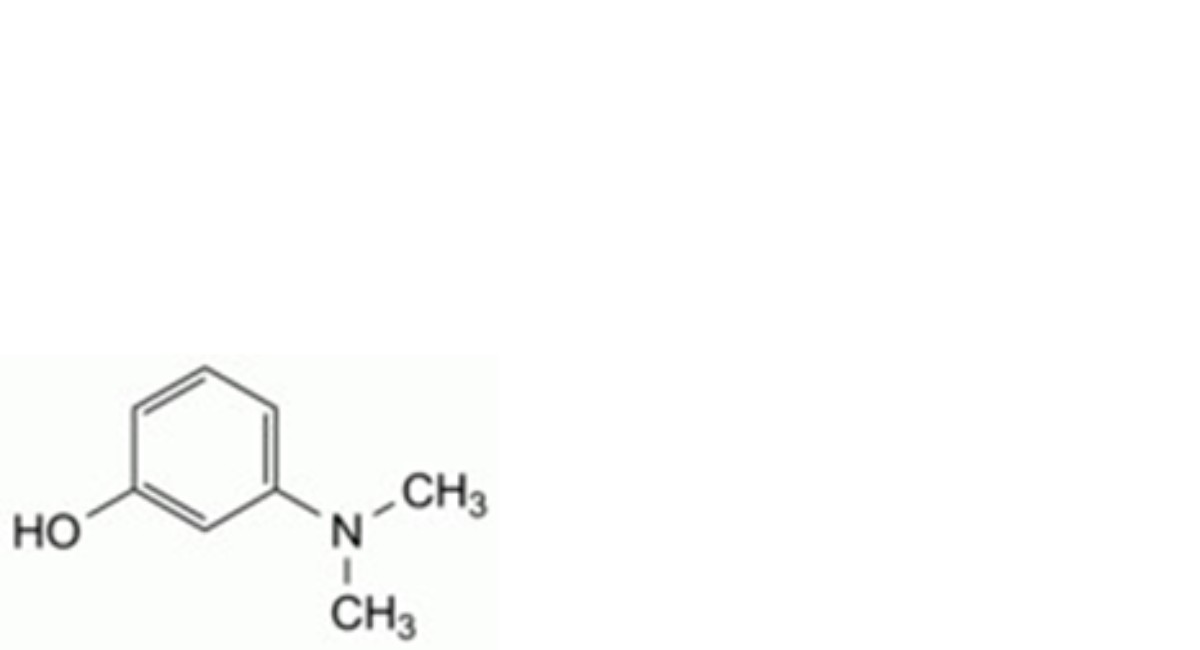
B. 3-(dimethylamino)phenol,
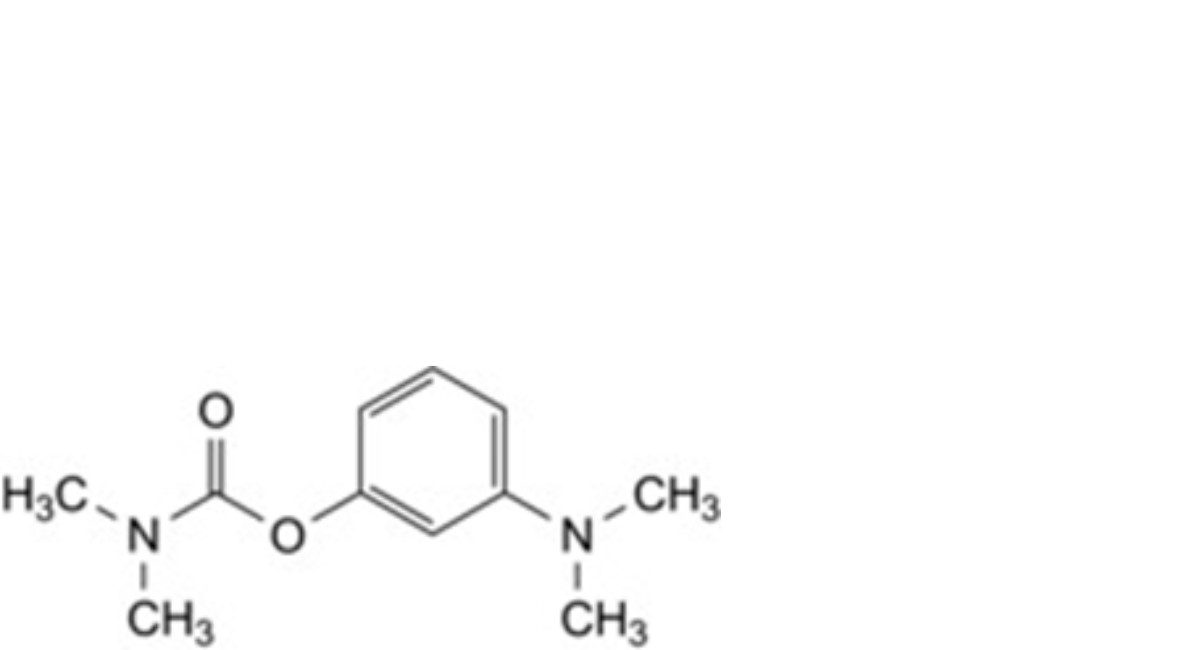
C. 3-(dimethylamino)phenyl dimethylcarbamate.
Ph Eur


