


Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Low molecular weight heparin.
Ph Eur
DEFINITION
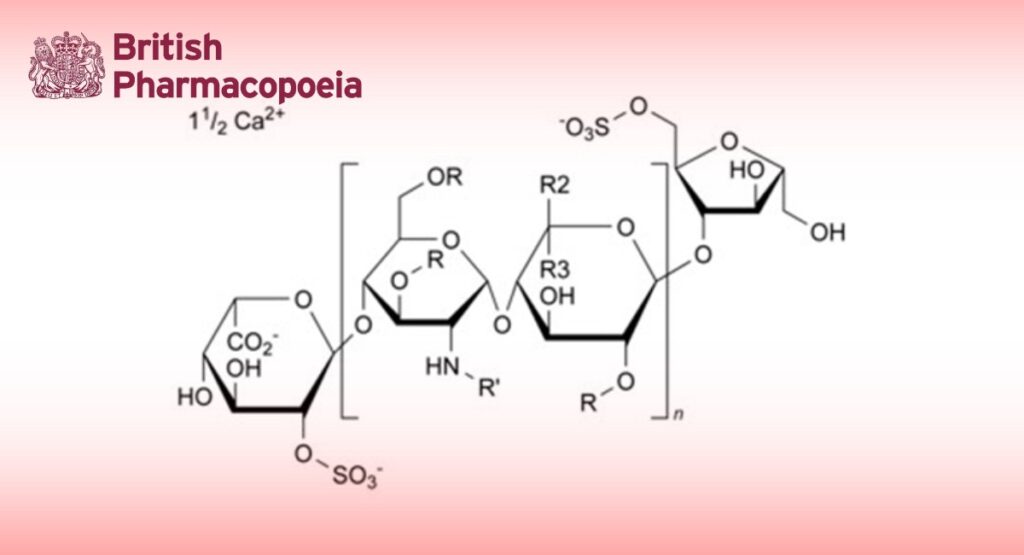
Calcium salt of low-molecular-mass heparin obtained by nitrous acid depolymerisation of heparin from pork intestinal mucosa, followed by fractionation to eliminate selectively most of the chains with a molecular mass lower than 2000. The majority of the components have a 2-O-sulfo-α-L-idopyranuronic acid structure at the non-reducing end and a 6-O-sulfo-2,5-anhydro-D-mannitol structure at the reducing end of their chain.
Nadroparin calcium complies with the monograph Low-molecular-mass heparins (0828) with the modifications and additional requirements below.
The mass-average relative molecular mass ranges between 3950 and 5350 with a characteristic value of about 4650.
The degree of sulfatation is about 2 per disaccharide unit.
The potency is not less than 95 IU and not more than 130 IU of anti-factor Xa activity per milligram, calculated with reference to the dried substance. The ratio of anti-factor Xa activity to anti-factor IIa activity is between 2.5 and 4.0.
IDENTIFICATION
Carry out identification test A as described in the monograph Low-molecular-mass heparins (0828) using nadroparin calcium CRS.
Carry out identification test C as described in the monograph Low-molecular-mass heparins (0828). The following requirements apply.
The mass-average relative molecular mass ranges between 3950 and 5350. The mass percentage of chains lower than 2000 is not more than 11 per cent. The mass percentage of chains between 2000 and 8000 ranges between 78 per cent and 98 per cent. The mass percentage of chains between 2000 and 4000 ranges between 32 per cent and 52 per cent.
TESTS
Appearance of solution
The solution is not more opalescent than reference suspension II (2.2.1) and not more intensely coloured than reference solution Y5 (2.2.2, Method II).
Dissolve 0.5 g in water R and dilute to 10 mL with the same solvent.
Ethanol
Head-space gas chromatography (2.2.28).
Internal standard solution Dilute 1.0 mL of 2-propanol R to 100.0 mL with water R. Dilute 1.0 mL of the solution to 50.0 mL with water R.
Blank solution 1.0 mL of water R.
Test solution (a) To 10.0 mg of the substance to be examined, add 1.0 mL of water R.
Test solution (b) To 10.0 mg of the substance to be examined, add 0.50 mL of water R and 0.50 mL of the internal standard solution.
Reference solution (a) Dilute 1.0 mL of anhydrous ethanol R to 100.0 mL with water R. Dilute 0.5 mL of the solution to 20.0 mL with water R.
Reference solution (b) To 0.50 mL of reference solution (a), add 0.50 mL of the internal standard solution.
Column:
— material: nickel;
— size: l = 1.5 m, Ø = 2 mm;
— stationary phase: ethylvinylbenzene-divinylbenzene copolymer R (150-180 µm).
Carrier gas helium for chromatography R or nitrogen for chromatography R. Flow rate 30 mL/min.
Static head-space conditions that may be used:
— equilibration temperature: 90 °C;
— equilibration time: 15 min;
— pressurisation time: 1 min.
Temperature:
— column: 150 °C;
— injection port and detector: 250 °C.
Detection Flame ionisation.
Identification of peaks Use the chromatogram obtained with reference solution (b) to identify the peaks due to ethanol and 2-propanol.
Retention time Ethanol = about 2.5 min; 2-propanol = about 4 min.
Calculate the percentage content m/m of ethanol taking its density at 20 °C to be 0.792 g/mL.
Limit:
— ethanol: maximum 1.0 per cent m/m.
N-NO groups
Maximum 0.25 ppm.
The content of N-NO-groups is determined by cleavage of the N-NO bond with hydrobromic acid in ethyl acetate under a reflux condenser and detection of the released NO by chemiluminescence.
Description of the apparatus (Figure 1134.-1). Use a 500 mL borosilicate glass round-bottomed flask, above which is attached a condenser which is equipped with:
— on one side, a torion joint through which a stream of argon R can be introduced via a cannula;
— on the other side, a screw joint with a piston equipped with a septum through which the reference solution and test solution will be injected.
The round-bottomed flask is connected in series to 3 bubble traps which are themselves connected to 2 cold traps, which are in turn connected to a chemiluminescence detector. Suitable tubing ensures the junctions are leak-free.
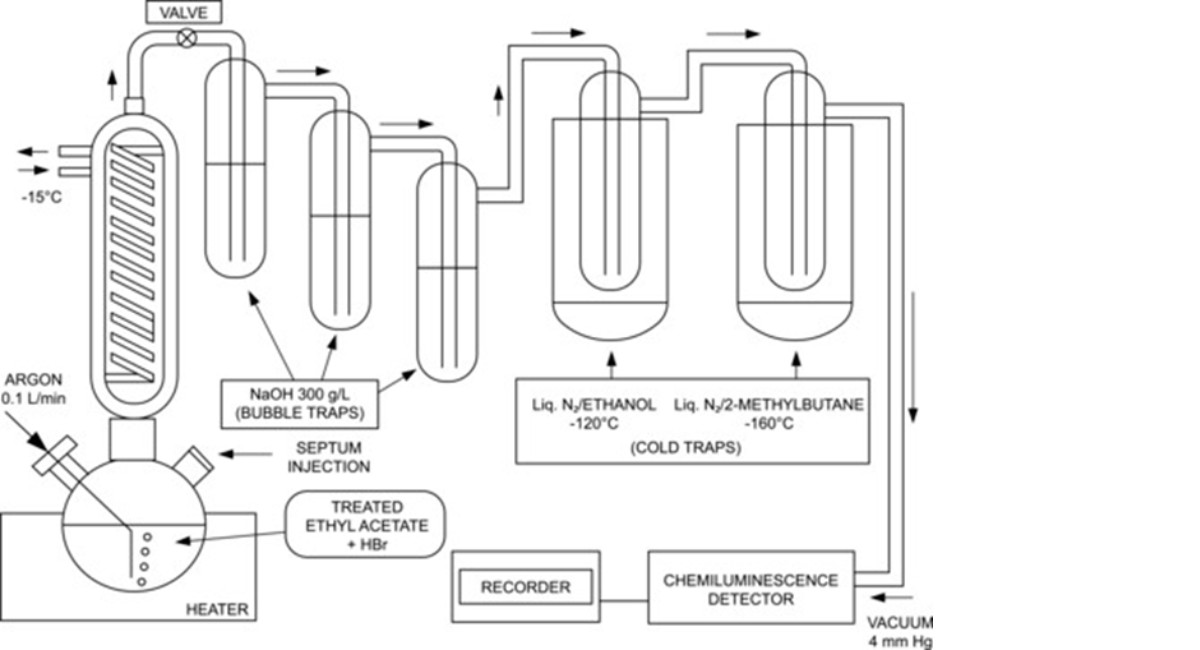
Flask: round-bottom borosilicate glass flask equipped with a central rodavis joint, a torion joint on the left neck and a 15 mm screw joint on the right neck; septum: silicone material, diameter 14 mm and thickness 3.5 mm.
Condenser: height 21 cm and internal diameter 3 cm, with a lower rodavis joint and an upper torion joint.
Bubble traps: height 24 cm and internal diameter 2.5 cm; internal tubing: length 23 cm and internal diameter 0.5 cm. Equipped with a centrally positioned rotulex mounting with torion joints on the inlet and outlet.
Cold traps: height 16.5 cm and internal diameter 4 cm; internal tubing: length 14 cm and internal diameter 1.3 cm. Equipped with torion joints on the inlet and outlet and placed in an isothermic flask: internal depth 22 cm and internal diameter 8 cm.
Tubing: fluorinated ethylene propylene material, internal diameter 3.2 mm and thickness 0.8 mm.
Figure 1134.-1. – Apparatus used for the assay of N-NO groups
Preparation of the chemiluminescence detector Switch on the chemiluminescence detector 48 h before use and start the vacuum pump. The vacuum must be less than 0.5 mm Hg. 1 h before use, open the oxygen valve at a pressure of 0.2 MPa and a flow rate of 9.4 mL/min.
Preparation of the bubble trap In each bubble trap, place 30 mL of a 300 g/L solution of sodium hydroxide R in water R.
Preparation of the cold traps.
— Trap at -120 °C: Slowly add liquid nitrogen to an isothermic flask containing 250 mL of anhydrous ethanol R whilst stirring with a wooden spatula until a paste is obtained. Place the cold trap in the isothermic flask prepared as described.
— Trap at -160 °C: Slowly add liquid nitrogen to an isothermic flask containing 250 mL of 2- methylbutane R whilst stirring with a wooden spatula until a paste is obtained. Place the cold trap in the isothermic flask prepared as described.
Drying of the 500 mL borosilicate-glass round-bottomed flask and condenser Boil 50 mL of ethyl acetate R under reflux for 1 h under argon R without connecting the system to the chemiluminescence detector.
Test solution Dry the substance to be examined for 12 h over diphosphorus pentoxide R at 60 °C under vacuum. Dissolve 0.10 g of the treated substance to be examined in 1.0 mL of treated formamide R. Shake the solution obtained for 30 min.
Reference solution Dilute 0.1 mL of nitrosodipropylamine solution R with 6.0 mL of anhydrous ethanol R. Dilute 0.1 mL of the solution obtained with 1.0 mL of treated formamide R. (This solution is equivalent to 0.05 ppm of N-NO groups).
Place 50 mL of treated ethyl acetate R in the dry 500 mL borosilicate glass round-bottomed flask equipped with a septum. Connect the round-bottomed flask to the condenser which has been previously cooled to -15 °C for 2 h.
Connect the argon R cannula and adjust the flow rate to 0.1 L/min. Check that the system is leak-free. Only the connector to the chemiluminescence detector remains open in order to avoid excess pressure.
Heat the treated ethyl acetate R to boiling.
Evacuate the system by slowly turning the valve of the chemiluminescence detector. At the same time tighten the inlet on the chemiluminescence detector.
When the system is equilibrated, the vacuum reaches 4 mm Hg.
The signal of the zero adjuster on the chemiluminescence detector is set to 10 per cent of the full scale of the recorder.
Through the septum of the 500 mL borosilicate glass round-bottomed flask, sequentially inject 0.5 mL of water R, 2.0 mL of dilute hydrobromic acid R and then another 2.0 mL of dilute hydrobromic acid R, making sure that the recorder pen has returned to the baseline between each injection.
Inject 50.0 µL of the reference solution, then 50.0 µL of the test solution after the recorder pen has returned to the baseline.
Calculate the content of N-NO groups of the substance to be examined.
Free sulfates
Liquid chromatography (2.2.29).
Test solution Dissolve 30.0 mg of the substance to be examined in water R and dilute to 10.0 mL with the same solvent.
Reference solution Dissolve 1.4787 g of anhydrous sodium sulfate R in water R and dilute to 1000.0 mL with the same solvent. Dilute 1.0 mL of the solution to 200.0 mL with distilled water R (5 ppm of sulfate ions).
Column:
— size: l = 50 mm, Ø = 4.6 mm;
— stationary phase: anion-exchange resin.
Chemical neutralisation system Neutralisation micromembrane in line with the mobile phase for anion detection; continuously pump in counter-flow with a 2.45 g/L solution of sulfuric acid R, at a flow rate of 4 mL/min.
Mobile phase:
— mobile phase A: 1.91 g/L solution of disodium tetraborate R;
— mobile phase B: 0.1 M sodium hydroxide;
| Time (min) | Mobile phase A (per cent V/V) | Mobile phase B (per cent V/V) |
| 0 – 15 | 100 | 0 |
| 15 – 15.5 | 100 → 0 | 0 → 100 |
| 15.5 – 25.5 | 0 | 100 |
Flow rate 1.0 mL/min.
Detection Conductivity detector with a sensitivity of 30 µS.
Injection 50 µL.
Identification of peaks Use the chromatogram obtained with the reference solution to identify the principal peak due to the sulfate ion.
Retention time Sulfate ion = about 7.5 min. Change the composition of the mobile phase, if necessary, to obtain the prescribed retention time.
Limit:
— free sulfates: maximum 0.5 per cent.
Ph Eur


