



Edition: BP 2025 (Ph. Eur. 11.6 update)
Action and use
Fluoroquinolone antibacterial.
DEFINITION
Levofloxacin Tablets contain Levofloxacin Hemihydrate.
The Tablets comply with the requirements stated under Tablets and with the following requirements.
Content of levofloxacin, C18H20FN3O4
95.0 to 105.0% of the stated amount.
IDENTIFICATION
In the Assay, record the UV spectrum of the principal peak in the chromatograms obtained with solutions (1) and (2) with a diode array detector in the range of 210 to 400 nm.
The UV spectrum of the principal peak in the chromatogram obtained with solution (1) is concordant with that of the peak in the chromatogram obtained with solution (2);
the retention time of the principal peak in the chromatogram obtained with solution (1) is similar to that of the peak in the chromatogram obtained with solution (2).
TESTS
Dissolution
Comply with the dissolution test for tablets and capsules, Appendix XII B1.
TEST CONDITIONS
(a) Use Apparatus 1, rotating the basket at 100 revolutions per minute.
(b) Use 900 mL of 0.1M hydrochloric acid, at a temperature of 37°, as the medium.
PROCEDURE
(1) After 30 minutes withdraw a sample of the medium and measure the absorbance of the filtered sample, suitably diluted with the dissolution medium, if necessary, to produce a solution expected to contain the equivalent of 0.027% w/v of levofloxacin, at the maximum at 293nm, Appendix II B, using dissolution medium in the reference cell.
(2) Measure the absorbance of a 0.027% w/v solution of levofloxacin hemihydrate BPCRS in the dissolution medium using dissolution medium in the reference cell.
DETERMINATION OF CONTENT
Calculate the total content of levofloxacin, C18H20FN3O4 in the medium from the absorbances obtained and using the declared content of C18H20FN3O4,1⁄2H2O, in levofloxacin hemihydrate BPCRS. Each mg of C18H20FN3O4,1⁄2H2O is equivalent to 0.9757 mg of C18H20FN3O4.
LIMITS
The amount of levofloxacin released is not less than 75% (Q) of the stated amount.
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions in the mobile phase.
(1) Disperse a quantity of the powdered tablets containing the equivalent of 0.5 g of levofloxacin with 75 mL of 20% v/v of acetonitrile. Dilute with 20% v/v of acetonitrile to produce 100 mL and filter. Dilute 1 volume to 10 volumes.
(2) Dilute 1 volume of solution (1) to 200 volumes.
(3) Dissolve the contents of a vial of levofloxacin for system suitability EPCRS in 1 mL.
(4) Dilute 1 volume of solution (2) to 5 volumes.
CHROMATOGRAPHIC CONDITIONS
(a) Use a stainless steel column (25 cm × 4.6 mm) packed with base-deactivated end-capped octadecylsilyl silica gel for chromatography (5 μm) (Inertsil ODS is suitable).
(b) Use isocratic elution and the mobile phase described below.
(c) Use a flow rate of 0.8 mL per minute.
(d) Use a column temperature of 45°.
(e) Use a detection wavelength of 360 nm.
(f) Inject 25 μL of each solution.
(g) Allow the chromatography to proceed for three times the retention time of levofloxacin.
MOBILE PHASE
0.0875% w/v of copper sulphate pentahydrate, 0.091% w/v of isoleucine and 0.595% w/v of ammonium acetate in 30% v/v of methanol.
SYSTEM SUITABILITY
The test is not valid unless, in the chromatogram obtained with solution (3), the resolution between the peaks due to impurities B and G is at least 1.5.
CALCULATION OF IMPURITIES
For all impurities, use the concentration of levofloxacin in solution (2).
For the reporting threshold, use the concentration of levofloxacin in solution (4).
For peak identification, use solution (3).
Levofloxacin retention time: about 20 minutes.
Relative retention: impurity E, about 0.4; impurity B, about 0.5; impurity G, about 0.56; impurity 1, about 0.63; impurity A, about 1.2.
Correction factors: impurity B, multiply by 1.3; impurity E, multiply by 1.7.
LIMITS
— impurity A: not more than 0.5%;
— unspecified impurities: for each impurity, not more than 0.2%;
— total impurities: not more than 1.0%;
— reporting threshold: 0.1%.
ASSAY
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
(1) Disperse a quantity of the whole tablets containing the equivalent of 2.5 g of levofloxacin in 375 mL of 20% v/v of acetonitrile. Dilute with 20% v/v of acetonitrile to produce 500 mL and filter. Dilute 1 volume to 25 volumes with the mobile phase.
(2) 0.2% w/v of levofloxacin hemihydrate BPCRS in 20% v/v of acetonitrile, dissolved with the aid of ultrasound if necessary. Dilute 1 volume to 10 volumes with the mobile phase.
CHROMATOGRAPHIC CONDITIONS
The chromatographic conditions described under Related substances may be used.
DETERMINATION OF CONTENT
Calculate the content of C18H20FN3O4 in the tablets using the declared content of C18H20FN3O4,1⁄2H2O in levofloxacin hemihydrate BPCRS. Each mg of C18H20FN3O4,1⁄2H2O is equivalent to 0.9757 mg of C18H20FN3O4.
LABELLING
The quantity of active ingredient is stated in terms of the equivalent amount of levofloxacin.
IMPURITIES
The impurities limited by the requirements of this monograph include impurities A, B, C, D, E, G, H and I listed under Levofloxacin Hemihydrate and:
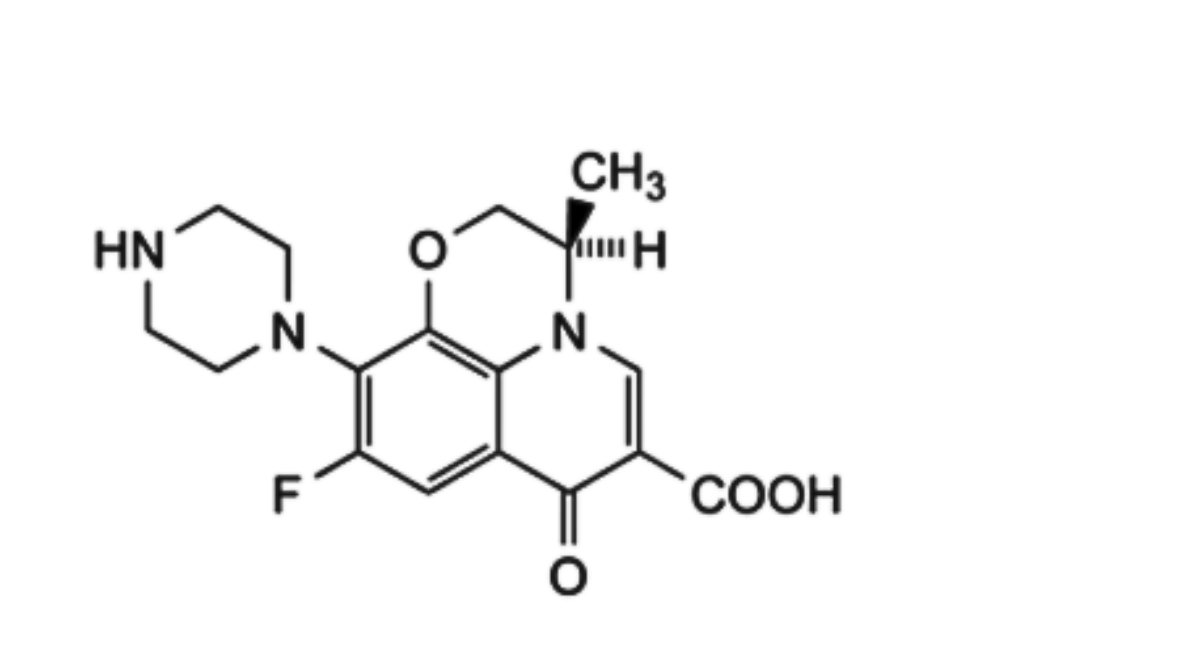
1. (S)-4-(6-Carboxy-9-fluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido-[1,2,3-de][1,4]benzoxazine-10-yl)-1-methylpiperazine 1-oxide (N-Oxide)


