


(Ph. Eur. monograph 2009)
C24H21I6N5O8 1269 59017-64-0
Action and use
Iodinated contrast medium.
DEFINITION
3-[[[[3-(Acetylmethylamino)-2,4,6-triiodo-5-(methylcarbamoyl)benzoyl]amino]acetyl]amino]-5-[(2- hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoic acid.
Content
98.5 per cent to 101.5 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white, hygroscopic powder.
Solubility
Very slightly soluble in water, slightly soluble in ethanol (96 per cent), very slightly soluble in methylene chloride. It dissolves in dilute solutions of alkali hydroxides.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Ioxaglic Acid
Comparison ioxaglic acid CRS.
TESTS
Appearance of solution
The solution is clear (2.2.1).
Dissolve 1.0 g in a 40 g/L solution of sodium hydroxide R and dilute to 20 mL with the same solution.
Absorbance (2.2.25)
Maximum 0.18, calculated for a solution containing 40 per cent of anhydrous ioxaglic acid.
Dissolve 10.0 g in about 8 mL of a 40 g/L solution of sodium hydroxide R. Adjust to pH 7.2-7.6 with a 40 g/L solution of sodium hydroxide R or 1 M hydrochloric acid. Dilute to 25 mL with water R. Filter through a membrane filter (nominal pore size 0.45 μm). Measure the absorbance at 450 nm using water R as the compensation liquid.
Related substances
Liquid chromatography (2.2.29): use the normalisation procedure.
Solvent mixture acetonitrile R, water R (5:95 V/V).
Test solution: Dissolve 0.10 g of the substance to be examined in about 40 mL of the solvent mixture. Add 0.5 ± 0.1 mL of a 4 g/L solution of sodium hydroxide R and dilute to 50.0 mL with the solvent mixture. Shake until dissolution is complete, using ultrasound if necessary.
Reference solution (a): Dissolve 0.10 g of ioxaglic acid CRS in about 40 mL of the solvent mixture. Add 0.5 ± 0.1 mL of a 4 g/L solution of sodium hydroxide R and dilute to 50.0 mL with the solvent mixture. Shake until dissolution is complete, using ultrasound if necessary.
Reference solution (b): Dissolve 5 mg of ioxaglic acid impurity A CRS in the solvent mixture and dilute to 50.0 mL with the solvent mixture. Dilute 1.0 mL of the solution to 50.0 mL with the solvent mixture.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: end-capped octylsilyl silica gel for chromatography R (5 μm);
— temperature: 25 °C.
Mobile phase:
— mobile phase A: 0.136 g/L solution of potassium dihydrogen phosphate R previously adjusted to pH 3.0 with phosphoric acid R;
— mobile phase B: acetonitrile R;
| Time
(min) |
Mobile phase A
(per cent V/V) |
Mobile phase B
(per cent V/V) |
| 0 – 5 | 95 → 90 | 5 → 10 |
| 5 – 40 | 90 | 10 |
| 40 – 85 | 90 → 70 | 10 → 30 |
| 85 – 115 | 70 | 30 |
| 115 – 120 | 70 → 50 | 30 → 50 |
| 120 – 125 | 50 | 50 |
Flow rate: 0.8 mL/min.
Detection: Spectrophotometer at 242 nm.
Injection: 10 μL.
Identification of impurities: Use the chromatogram supplied with ioxaglic acid CRS and the chromatogram obtained with reference solution (a) to identify the peaks due to impurities B, C, D1, D2, D3, D4, E and F; use the chromatogram obtained with reference solution (b) to identify the peak due to impurity A.
Relative retention: With reference to ioxaglic acid (retention time = about 65 min): impurity A = about 0.3; impurity B = about 0.7; impurity C = about 0.9; impurity D1 = about 1.09; impurity E = about 1.12; impurity D2 = about 1.20; impurity D3 = about 1.26; impurity D4 = about 1.28; impurity F = about 1.6.
System suitability: Reference solution (a):
— peak-to-valley ratio: minimum 1.3, where Hp = height above the baseline of the peak due to impurity C and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to ioxaglic acid. Limits:
— impurity D (sum of the peaks due to impurities D1, D2, D3 and D4): maximum 0.7 per cent;
— impurity E: maximum 0.7 per cent;
— impurity F: maximum 0.4 per cent;
— impurity B: maximum 0.3 per cent;
— impurity C: maximum 0.3 per cent;
— impurity A: maximum 0.1 per cent;
— unspecified impurities: for each impurity, maximum 0.2 per cent;
— total: maximum 2.0 per cent;
— reporting threshold: 0.05 per cent; disregard any peak with a retention time greater than 125 min.
The thresholds indicated under Related substances (Table 2034.-1) in the general monograph Substances for pharmaceutical use (2034) do not apply.
Iodides
Maximum 50 ppm.
Disperse 10.0 g in 50 mL of water R. Add 8 mL of 1 M sodium hydroxide. After dissolution and homogenisation, add 1.0 mL of glacial acetic acid R. Immediately titrate with 0.001 M silver nitrate, determining the end-point potentiometrically (2.2.20), using a silver indicator electrode and a suitable reference electrode.
1 mL of 0.001 M silver nitrate is equivalent to 0.1269 mg of iodides.
Water (2.5.12)
Maximum 5.0 per cent, determined on 0.100 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
In a round-bottomed flask place 0.100 g of the substance to be examined and add 5 mL of strong sodium hydroxide solution R, 20 mL of water R, 1 g of zinc powder R and a few glass beads. Fit the flask with a reflux condenser and boil for 30 min. Cool and rinse the condenser with 20 mL of water R. Add the rinsings to the contents of the flask. Filter, wash the filter with 3 quantities, each of 15 mL, of water R and add the washings to the filtrate. Add 40 mL of dilute sulfuric acid R and titrate immediately with 0.05 M silver nitrate. Determine the end-point potentiometrically (2.2.20).
1 mL of 0.05 M silver nitrate is equivalent to 10.58 mg of C24H21I6N5O8.
STORAGE
In an airtight container, protected from light.
IMPURITIES
Specified impurities A, B, C, D, E, F.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities. It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) G, H.
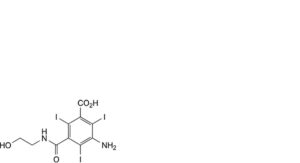
A. 3-amino-5-[(2-hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoic acid,
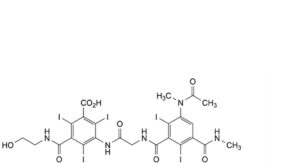
B. 3-[[[[3-(acetylmethylamino)-2,6-diiodo-5-(methylcarbamoyl)benzoyl]amino]acetyl]amino]-5-[(2- hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoic acid,
C. unknown structure,
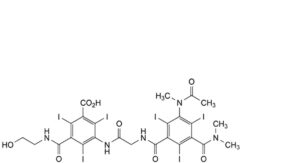
D. D1, D2, D3 and D4: 3-[[[[3-(acetylmethylamino)-5-(dimethylcarbamoyl)-2,4,6-triiodobenzoyl]amino]acetyl]amino]-5-[(2-hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoic acid,
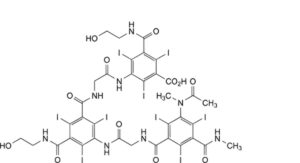
E. 3-[[[[3-[[[[3-(acetylmethylamino)-2,4,6-triiodo-5-(methylcarbamoyl)benzoyl]amino]acetyl]amino]-5-[(2- hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoyl]amino]acetyl]amino]-5-[(2-hydroxyethyl)carbamoyl]-2,4,6-triiodobenzoic acid,
F. unknown structure,
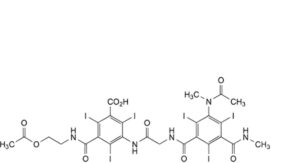
G. 3-[[[[3-(acetylmethylamino)-2,4,6-triiodo-5-(methylcarbamoyl)benzoyl]amino]acetyl]amino]-5-[[2-(acetyloxy)ethyl]carbamoyl]-2,4,6-triiodobenzoic acid,
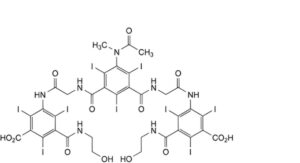
H. 3,3′-[[5-(acetylmethylamino)-2,4,6-triiodo-1,3-phenylene]bis(carbonyliminomethylenecarbonylimino)]bis[5-[(2-hydroxyethyl)carbamoyl]-2,4,6- triiodobenzoic] acid.


