


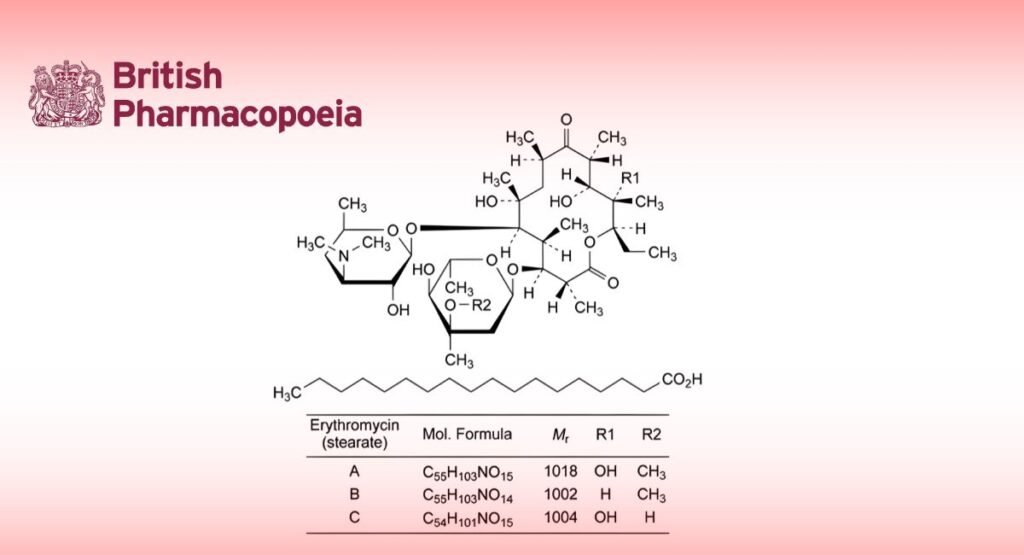
(Ph. Eur. monograph 0490)
Action and use
Macrolide antibacterial.
Preparation
Erythromycin Stearate Tablets
DEFINITION
Mixture of the stearate salts of erythromycin and stearic acid.
Main component (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-
hexopyranosyl)oxy]-14-ethyl-7,12,13-trihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione octadecanoate (erythromycin A stearate).
Salt of a product obtained by fermentation using a strain of Streptomyces erythreus.
Content
— sum of erythromycins A, B and C expressed as stearates: 79.0 per cent to 102.0 per cent (anhydrous substance);
— erythromycin B stearate: maximum 5.0 per cent (anhydrous substance);
— erythromycin C stearate: maximum 5.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Practically insoluble in water, soluble in acetone and in methanol.
Solutions may be opalescent.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparison erythromycin stearate CRS.
TESTS
Free stearic acid
Maximum 14.0 per cent (anhydrous substance) of C18H36O2.
Dissolve 0.400 g in 50 mL of methanol R. Titrate with 0.1 M sodium hydroxide, determining the end-point potentiometrically (2.2.20). Calculate the volume of 0.1 M sodium hydroxide required per gram of the substance to be examined (n1 mL).
Dissolve 0.500 g in 30 mL of methylene chloride R. If the solution is opalescent, filter and shake the residue with 3 quantities, each of 25 mL, of methylene chloride R. Filter, if necessary, and rinse the filter with methylene chloride R.
Reduce the volume of the combined filtrate and rinsings to 30 mL by evaporation on a water-bath. Add 50 mL of glacial acetic acid R and titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20). Calculate the volume of 0.1 M perchloric acid required per gram of the substance to be examined (n2 mL).
Calculate the percentage content of C18H36O2 using the following expression:
2.845(n1-n2)x100/(100-h)
h = percentage water content.
Related substances
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Solution A: Dissolve 11.5 g of dipotassium hydrogen phosphate R in 900 mL of water R, adjust to pH 8.0 with dilute phosphoric acid R and dilute to 1000 mL with water R.
Solvent mixture methanol R, solution A (40:60 V/V).
Test solution: Dissolve 55.0 mg of the substance to be examined in the solvent mixture and dilute to 10.0 mL with the solvent mixture. Centrifuge and use the clear solution.
Reference solution (a): Dissolve 40.0 mg of erythromycin A CRS in the solvent mixture and dilute to 10.0 mL with the solvent mixture.
Reference solution (b): Dissolve 10.0 mg of erythromycin B CRS and 10.0 mg of erythromycin C CRS in the solvent mixture and dilute to 50.0 mL with the solvent mixture.
Reference solution (c): Dilute 1.0 mL of reference solution (a) to 100.0 mL with the solvent mixture.
Reference solution (d): Dissolve 4 mg of erythromycin for system suitability CRS (containing impurities A, B, C, D, E, F, H and L) in the solvent mixture and dilute to 1 mL with the solvent mixture.
Reference solution (e): Dissolve 4 mg of erythromycin stearate for impurity S identification CRS in the solvent mixture and dilute to 1 mL with the solvent mixture.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: end-capped polar-embedded octadecylsilyl amorphous organosilica polymer R (3.5 μm);
— temperature: 65 °C; preheating the mobile phase may be required, for instance by extending the inlet tubing in the oven to 30 cm.
Mobile phase:
— mobile phase A: phosphate buffer solution pH 7.0 R7, acetonitrile R1, water for chromatography R (5:35:60 V/V/V);
— mobile phase B: phosphate buffer solution pH 7.0 R7, water for chromatography R, acetonitrile R1 (5:45:50 V/V/V);
| Time (min) |
Mobile phase A (per cent V/V) |
Mobile phase B (per cent V/V) |
| 0 – tR | 100 | 0 |
| tR – (tR + 2) | 100 → 0 | 0 → 100 |
| (tR + 2) – (tR + 15) | 0 | 100 |
tR = retention time of erythromycin B, determined by injecting 10 μL of reference solution (b) and eluting with mobile phase A
Flow rate 1.0 mL/min.
Detection: Spectrophotometer at 210 nm.
Autosampler Set at 4 °C.
Injection 100 μL.
Identification of impurities: Use the chromatogram supplied with erythromycin for system suitability CRS and the chromatogram obtained with reference solution (d) to identify the peaks due to impurities A, B, C, D, E, F and L; use the chromatogram supplied with erythromycin stearate for impurity S identification CRS and the chromatogram obtained with reference solution (e) to identify the peak due to impurity S; use the chromatogram obtained with reference solution (b) to identify the peaks due to erythromycins B and C.
Relative retention With reference to erythromycin A (retention time = about 23 min): impurity A = about 0.4;
impurity B = about 0.5; erythromycin C = about 0.55; impurity L = about 0.63; impurity C = about 0.9;
impurity D = about 1.61; erythromycin B = about 1.75; impurity F = about 1.81; impurity S = about 2.1; impurity E = about 2.3.
System suitability Reference solution (d):
— resolution: minimum 1.2 between the peaks due to impurity B and erythromycin C;
— peak-to-valley ratio: minimum 1.5, where Hp = height above the baseline of the peak due to impurity F and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to erythromycin B; minimum 2.0, where Hp = height above the baseline of the peak due to impurity C and Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to erythromycin A. If necessary, adjust the concentration of acetonitrile R1 in the mobile phases and/or the gradient to obtain the required separation.
Calculation of percentage contents:
— correction factors: multiply the peak areas of the following impurities by the corresponding correction factor: impurity D = 2; impurity E = 0.08; impurity F = 0.08; impurity L = 0.11;
— for each impurity, use the concentration of erythromycin A in reference solution (c).
Limits:
— impurity C: maximum 3.0 per cent;
— impurities A, B: for each impurity, maximum 2.0 per cent;
— impurities D, E, F, S: for each impurity, maximum 1.0 per cent;
— impurity L: maximum 0.4 per cent;
— any other impurity: for each impurity, maximum 0.4 per cent;
— total: maximum 7.0 per cent;
— reporting threshold: 0.2 per cent; disregard the peaks due to erythromycins B and C.
Water (2.5.12)
Maximum 4.0 per cent, determined on 0.300 g.
Use a 100 g/L solution of imidazole R in anhydrous methanol R as the solvent.
Sulfated ash (2.4.14)
Maximum 0.5 per cent, determined on 1.0 g.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modifications.
Injection Test solution and reference solutions (a) and (b).
System suitability Reference solution (a):
— symmetry factor: maximum 2.5 for the peak due to erythromycin A;
— repeatability: maximum relative standard deviation of 1.0 per cent determined on 6 injections.
Calculate the percentage content of erythromycin A (C37H67NO13) using the chromatogram obtained with reference solution (a). Calculate the percentage contents of erythromycin B (C37H67NO12) and erythromycin C (C36H65NO13) using the chromatogram obtained with reference solution (b).
Express the results as erythromycin A stearate, erythromycin B stearate and erythromycin C stearate by multiplying the percentage content of erythromycin A by 1.3869, the percentage content of erythromycin B by 1.3955 and the percentage content of erythromycin C by 1.3944.
For the calculation of content of erythromycin stearate, use the sum of erythromycins A, B and C expressed as stearates as described above.
STORAGE
Protected from light.
IMPURITIES
Specified impurities A, B, C, D, E, F, L, S.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities. It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) H, I, J, K, M, N.
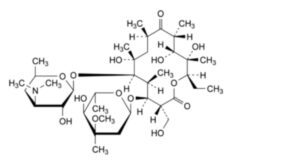
A. (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12,13-trihydroxy-3-(hydroxymethyl)-5,7,9,11,13-pentamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione (erythromycin F),
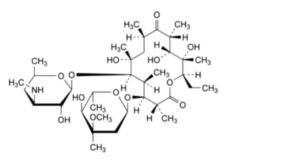
B. (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12,13-trihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(methylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione (3′′-N-demethylerythromycin A),
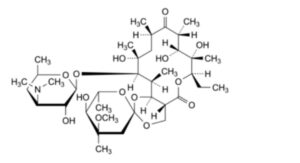
C. (2S,4aR,4′R,5′S,6′S,7R,8S,9R,10R,12R,14R,15R,16S,16aS)-7-ethyl-5′,8,9,14-tetrahydroxy-4′-methoxy-
4′,6′,8,10,12,14,16-heptamethyl-15-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-
hexopyranosyl]oxy]hexadecahydrospiro[5H,11H-1,3-dioxino[5,4-c]oxacyclotetradecin-2,2′-pyrane]-5,11-dione (erythromycin E),
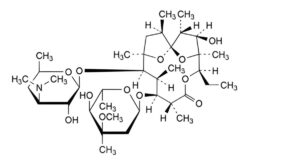
D. (1S,2R,3R,4S,5R,8R,9S,10S,11R,12R,14R)-9-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-5-ethyl-3-hydroxy-2,4,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-6,15,16-trioxatricyclo[10.2.1.1 ]hexadecan-7-one (anhydroerythromycin A),
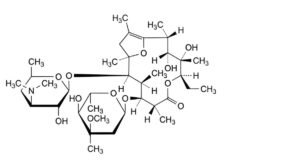
E. (2R,3R,4S,5R,8R,9S,10S,11R,12R)-9-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-5-ethyl-3,4-dihydroxy-2,4,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-6,15-dioxabicyclo[10.2.1]pentadec-1(14)-en-7-one (erythromycin A enol ether),
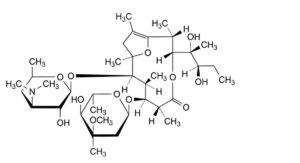
F. (2R,3R,6R,7S,8S,9R,10R)-7-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-3-[(1R,2R)-1,2-dihydroxy-1-methylbutyl]-2,6,8,10,12-pentamethyl-9-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-4,13-dioxabicyclo[8.2.1]tridec-1(12)-en-5-one (pseudoerythromycin A enol ether),
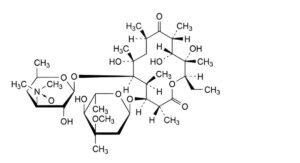
H. (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12,13-trihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione N-oxide (erythromycin A 3′′-N-oxide),
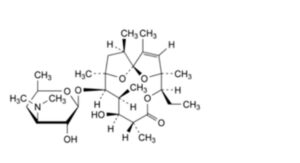
I. (1S,4S,5R,8R,9S,10S,11R,12R,14R)-5-ethyl-9-hydroxy-2,4,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-
(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-6,15,16-trioxatricyclo[10.2.1.1 ]hexadec-2-en-7-one (erythralosamine),
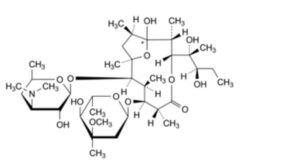
J. (1RS,2R,3R,6R,7S,8S,9R,10R,12R)-7-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-3-[(1R,2R)-1,2-dihydroxy-1-methylbutyl]-1-hydroxy-2,6,8,10,12-pentamethyl-9-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-4,13-dioxabicyclo[8.2.1]tridecan-5-one (pseudoerythromycin A hemiketal),
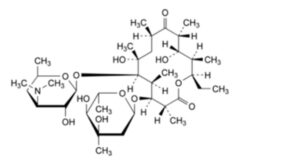
K. (3R,4S,5S,6R,7R,9R,11R,12S,13R,14R)-4-[(2,6-dideoxy-3-C-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12-dihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-
hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione (erythromycin D),
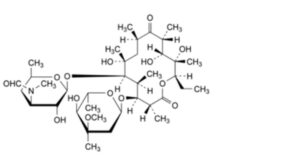
L. (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12,13-trihydroxy-3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(formylmethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione (3′′-N-demethyl-3′′-N-formylerythromycin A),
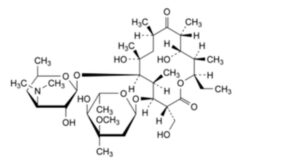
M. (3R,4S,5S,6R,7R,9R,11R,12S,13R,14R)-4-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-14-ethyl-7,12-dihydroxy-3-(hydroxymethyl)-5,7,9,11,13-pentamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]oxacyclotetradecane-2,10-dione (erythromycin G),
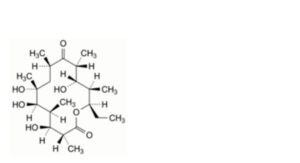
N. (3R,4S,5S,6R,7R,9R,11R,12S,13R,14R)-14-ethyl-4,6,7,12-tetrahydroxy-3,5,7,9,11,13-
hexamethyloxacyclotetradecane-2,10-dione (erythronolide B),
S. unknown structure.


