



Edition: BP 2025 (Ph. Eur. 11.6 update)
General Notices
Action and use
Low molecular weight heparin.
DEFINITION
Enoxaparin Sodium Injection is a sterile solution of Enoxaparin Sodium in Water for Injections. It may contain, in multidose containers, a suitable antimicrobial preservative.
PRODUCTION
The product is manufactured by validated methods to prevent the introduction of hypotensive substances and contamination by over-sulfated glycosaminoglycans. The method ensures compliance with the test for related substances.
The injection complies with the general requirements for Parenteral Preparations and the following specific tests.
Activity
— Anti-factor Xa activity: 90–110% of stated value.
— Ratio of anti-factor Xa to anti-factor IIa: 3.3–5.3.
IDENTIFICATION
A. Size-exclusion chromatography (Appendix III C).
(1) Dilute the injection to contain 1000 IU of anti-factor Xa per mL.
(2) 1% w/v heparin low-molecular-mass for calibration EPCRS.
CHROMATOGRAPHIC CONDITIONS
(a) Use a column (30 cm × 7.8 mm) packed with appropriate porous silica beads (5 μm) with a fractionation range for proteins of approximately 15 000 to 100 000 Da (Tosoh Bioscience TSK-GEL G2000SWXL or TSK-GEL G3000SWXL are suitable).
(b) Use isocratic elution and the mobile phase described below.
(c) Use a flow rate of 0.5 mL per minute.
(d) Use an ambient column temperature.
(e) For detection use a differential refractometer (RI).
(f) Inject 25 μL of each solution.
To calibrate, inject 25 μL of the reference solution and record the chromatogram for a period of time, ensuring complete elution of sample, salt and solvent peaks.
Calculate the total area under the RI curve (ΣRI) by numerical integration over the range of interest (i.e. excluding salt and solvent peaks at the end of the chromatogram as well as the artefact peak (*), that if present, elutes at the same retention time as the peak that would be obtained after injection of sodium sulfate), that if present, elutes at the same retention time as the peak that would be obtained after injection of sodium sulfate) as shown in Figure 1.
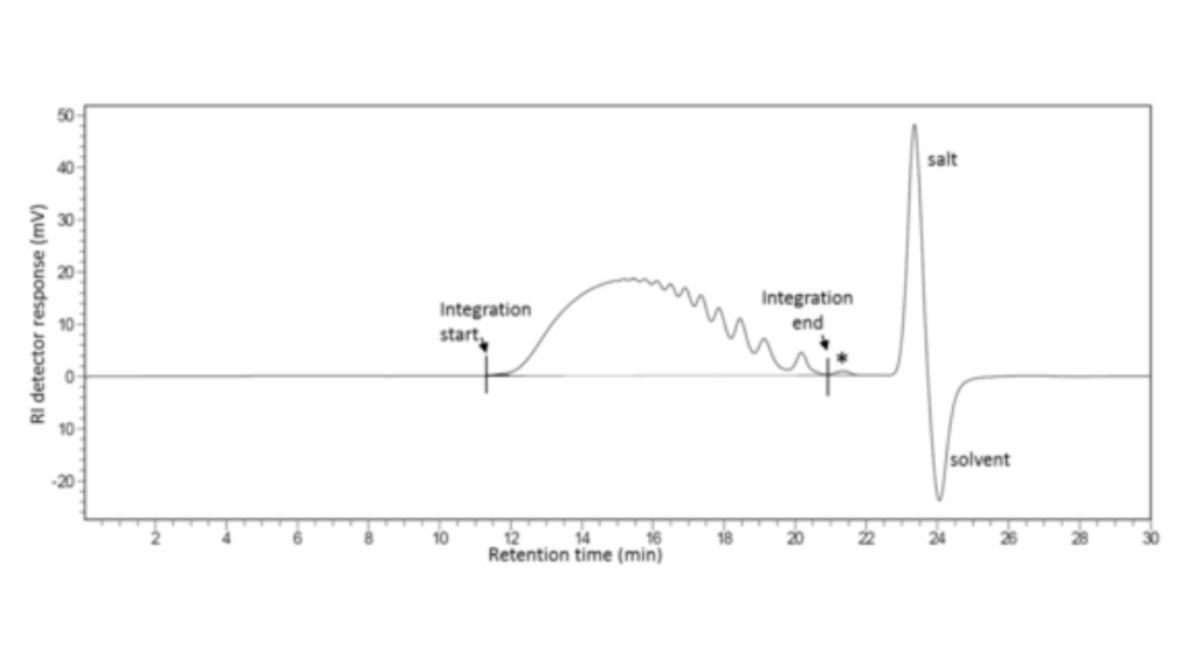
Calculate the area under the curve up to each point as a percentage of the total area.
Using the leaflet supplied with heparin low-molecular-mass for calibration EPCRS, identify those points in the chromatogram for which the percentage of the total area under the curve is closest to the percentage mass fractions listed in the Broad Standard Table, and assign the molecular mass in the table to the corresponding retention time in the chromatogram.
A calibration curve for the chromatographic system is derived by fitting a suitable mathematical relationship to the set of retention times and logarithms of the molecular masses.
A polynomial of the 3rd degree is recommended.
Inject 25 μL of the test solution and record the chromatogram for a sufficient period of time to ensure complete elution of sample, salt and solvent peaks.
The mass-average relative molecular mass is defined by the following expression:
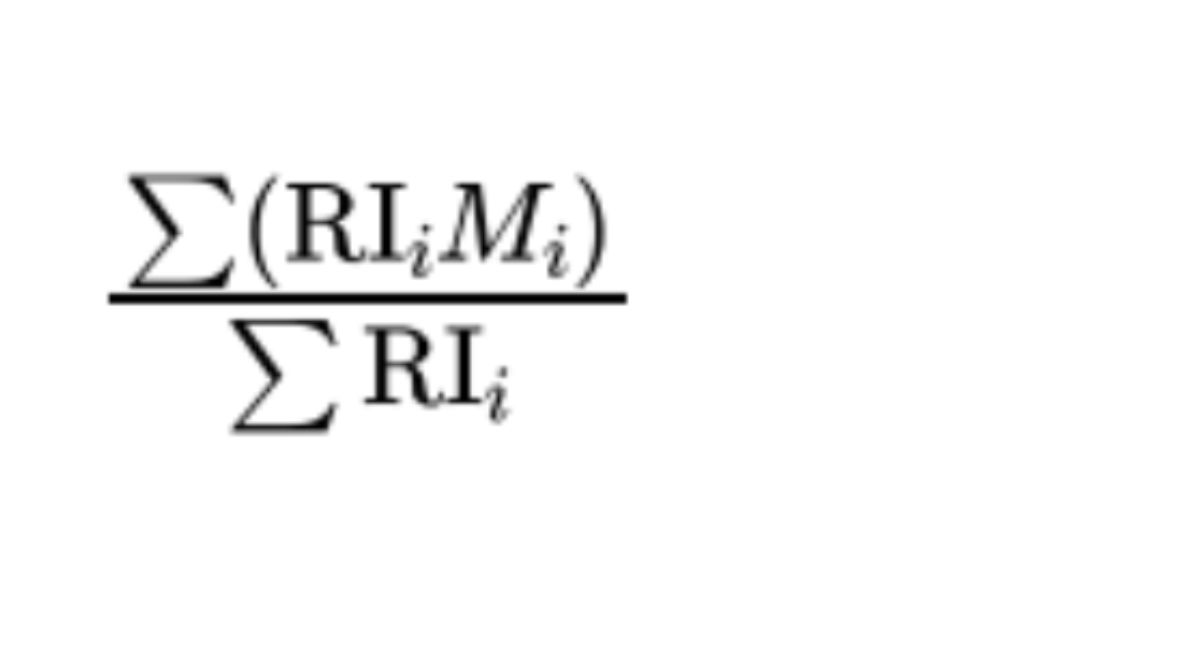
where:
RIᵢ = Refractive Index of substance eluting in the fraction i (proportional to the mass of the substance);
Mᵢ = relative molecular mass corresponding to fraction i.
MOBILE PHASE
0.77% w/v of ammonium acetate.
CONFIRMATION
The mass-average relative molecular mass ranges between 3800 and 5000.
The mass percentage of chains lower than 2000 is between 12.0 and 20.0%.
The mass percentage of chains between 2000 and 8000 ranges between 68.0 and 82.0%.
B. The ratio of anti-factor Xa activity to anti-factor IIa activity, determined as described under Assay, is not less than 3.3 and not more than 5.3.
C. Yields reaction A characteristic of sodium salts, Appendix VI.
TESTS
Clarity and colour of solution
The solution is clear, Appendix IV A, and not more intensely coloured than reference solution Y4 or BY4, Appendix IV B, Method II.
Acidity or alkalinity
pH, 5.5 to 7.5, Appendix V L.
Light absorption
The light absorption, Appendix II B, of a solution containing the total contents of a single-dose container, or 0.4 mL from a multi-dose container, diluted to 100.0 mL with a solution of 0.01M hydrochloric acid exhibits a maximum at 231 nm.
Related substances
Carry out the method for liquid chromatography, Appendix III D, using the following solutions.
Solutions (3) to (6) are stable at room temperature for 24 hours.
(1) Dilute the injection with water for chromatography to give 5000 IU per mL.
(2) Mix 500 µL of solution (1) and 250 µL of 1M hydrochloric acid, add 50 µL of a 25% w/v solution of sodium nitrite.
Mix gently and allow to stand at room temperature for 40 minutes. Add 200 µL of 1M sodium hydroxide to stop the reaction.
(3) Dissolve 0.25 g of heparin for physico-chemical analysis EPCRS in water for chromatography and dilute to 2.0 mL with the same solvent. Mix using a vortex mixer until dissolution is complete.
(4) Add 1200 µL of solution (3) to 300 µL of dermatan sulfate and over-sulfated chondroitin sulfate EPCRS. Mix using a vortex mixer to homogenise.
(5) Add 400 µL of solution (3) to 100 µL of water for chromatography and mix using a vortex mixer. Add 250 µL of 1M hydrochloric acid, then add 50 µL of a 25% w/v mL solution of sodium nitrite.
Mix gently and allow to stand at room temperature for 40 minutes before adding 200 µL of 1M sodium hydroxide to stop the reaction.
(6) To 500 µL of solution (4), add 250 µL of 1M hydrochloric acid, then add 50 µL of a 25% w/v solution of sodium nitrite.
Mix gently and allow to stand at room temperature for 40 minutes before adding 200 µL of 1M sodium hydroxide to stop the reaction.
CHROMATOGRAPHIC CONDITIONS
(a) Column: 25 cm × 2 mm packed with anion-exchange resin (9 µm) (Dionex Ionpac AS11-HC).
(b) Pre-column: 5 cm × 2 mm packed with anion-exchange resin (13 µm) (Dionex Ionpac AG11-HC).
(c) Flow rate: 0.22 mL/min.
(d) Column temperature: 40 °C.
(e) Detection: 202 nm.
(f) Injection: 20 µL of solutions (2), (5) and (6).
MOBILE PHASE
A: 0.040% w/v sodium dihydrogen orthophosphate in water for chromatography adjusted to pH 3.0 with dilute orthophosphoric acid.
B: 0.040% w/v sodium dihydrogen orthophosphate and 14% w/v sodium perchlorate in water for chromatography, adjusted to pH 3.0 with dilute orthophosphoric acid.
Equilibrate the column with the mobile phase in the initial gradient ratio for at least 15 minutes.
| Time (Minutes) | Mobile phase A (% v/v) | Mobile phase B (% v/v) | Comment |
| 0-10 | 76 | 25 | isocratic |
| 10-35 | 75→0 | 25→100 | linear gradient |
| 35-40 | 0 | 100 | isocratic |
| 40-45 | 0→75 | 100→25 | linear gradient |
| 45-60 | 75 | 25 | re-equilibration |
When the chromatograms are recorded under the prescribed conditions, the relative retention times with reference to heparin (retention time about 26 minutes) are dermatan sulfate and chondroitin sulfate ≈ 0.9 over-sulfated chondroitin sulfate ≈ 1.3
SYSTEM SUITABILITY
The test is not valid unless:
in the chromatogram obtained with solution (5), no peak is present at the retention time of heparin;
in the chromatogram obtained with solution (6), the resolution between dermatan/chondroitin sulfate and over-sulfated chondroitin sulfate ≥ 3.0.
LIMITS
In the chromatogram obtained with solution (2):
the area of the peak due to dermatan sulfate and chondroitin sulfate ≤ half the area of the corresponding peak in solution (6) (2.0%);
no peaks other than dermatan/chondroitin sulfate detected.
Sodium
10.2 to 16.9% of Enoxaparin Sodium, determined by atomic absorption spectrophotometry, Appendix II D Method I. For the purposes of this test, assume that 100 IU of anti-factor Xa is equivalent to 1 mg of Enoxaparin Sodium.
Bacterial endotoxins
Carry out the test for bacterial endotoxins, Appendix XIV C. The endotoxin limit concentration is less than 0.01 IU per International Unit of anti-Xa activity.
ASSAY
The anticoagulant activity of low-molecular-mass heparins is determined in vitro by two assays which determine its ability to accelerate the inhibition of factor Xa (anti-Xa assay) and thrombin, factor IIa (anti-IIa assay), by antithrombin III.
The International Units for anti-Xa and anti-IIa activity are the activities contained in a stated amount of the International Standard for low-molecular-mass heparin.
Heparin low-molecular-mass for assay EPBRP, calibrated in International Units by comparison with the International Standard using the two assays given below, is used as the reference preparation.
Provided the equivalence with the below methods is demonstrated, the volumes below can be adjusted in order to use automated methods.
For anti-factor Xa activity
Test solutions (1) to (4) Prepare a series of 4 independent dilutions of the injection being examined in tris-chloride buffer pH 7.4; the concentration range of the solutions should be within 0.025 IU to 0.2 IU of anti-factor Xa activity per mL and the dilutions chosen should give a linear response when results are plotted as absorbance against log concentration.
Reference solutions (1) to (4) Prepare a series of 4 dilutions of the reference preparation of low-molecular-mass heparin in tris-chloride buffer pH 7.4; the concentration range of the solutions should be within 0.025 IU to 0.2 IU of anti-factor Xa activity per mL and the dilutions chosen should give a linear response when results are plotted as absorbance against log concentration.
Label 16 tubes in duplicate: T1, T2, T3, T4 for the dilutions of the injection being examined and R1, R2, R3, R4 for the dilutions of the reference preparation. To each tube add 50 μL of antithrombin III solution R1 and 50 μL of the appropriate dilution of the injection being examined, or the reference preparation. After each addition, mix but do not allow bubbles to form. Treating the tubes in the order R1, R2, R3, R4, T1, T2, T3, T4, T1, T2, T3, T4, R1, R2, R3, R4, allow to equilibrate at 37° (in a water-bath or heating block) for 1 minute and add to each tube 100 μL of bovine factor Xa solution. Incubate for exactly 1 minute and add 250 μL of chromophore substrate R1. Stop the reaction after exactly 4 minutes by adding 375 μL of acetic acid. Transfer the mixtures to semi-micro cuvettes and measure the absorbance at 405 nm, Appendix II B, using a suitable reading device. Determine the blank amidolytic activity at the beginning and at the end of the procedure in a similar manner, using tris-chloride buffer pH 7.4 in place of the reference and test solutions; the 2 blank values do not differ significantly. Calculate the regression of the absorbance on log concentrations of the solutions of the injection being examined and of the reference preparation of low-molecular-mass heparins and calculate the potency of the substance being examined in International Units of anti-factor Xa activity per mL using the usual statistical methods for parallel-line assays.
For anti-factor IIa activity
Test solutions (1) to (4) Prepare a series of 4 independent dilutions of the injection being examined in tris-chloride buffer pH 7.4; the concentration range should be within 0.015 IU to 0.075 IU of anti-factor IIa activity per mL and the dilutions chosen should give a linear response when results are plotted as absorbance against log concentration.
Reference solutions (1) to (4) Prepare a series of 4 independent dilutions of the reference preparation of low-molecular-mass heparin in tris-chloride buffer pH 7.4; the concentration range should be within 0.015 IU to 0.075 IU of anti-factor IIa activity per mL and the dilutions chosen should give a linear response when results are plotted as absorbance against log concentration.
Label 16 tubes, in duplicate: T1, T2, T3, T4 for the dilutions of the injection being examined and R1, R2, R3, R4 for the dilutions of the reference preparation. To each tube add 50 μL of antithrombin III solution R2 and 50 μL of the appropriate dilution of the injection being examined or the reference preparation. After each addition, mix but do not allow bubbles to form. Treating the tubes in the order R1, R2, R3, R4, T1, T2, T3, T4, T1, T2, T3, T4, R1, R2, R3, R4, allow to equilibrate at 37° (in a water-bath or heating block) for 1 minute and add to each tube 100 μL of human thrombin solution. Incubate for exactly 1 minute and add 250 μL of chromophore substrate R2. Stop the reaction after exactly 4 minutes by adding 375 μL of acetic acid. Transfer the mixtures to semi-micro cuvettes and measure the absorbance at 405 nm, Appendix II B, using a suitable reading device. Determine the blank amidolytic activity at the beginning and at the end of the procedure in a similar manner, using tris-chloride buffer pH 7.4 in place of the reference and test solutions; the 2 blank values do not differ significantly. Calculate the regression of the absorbance on log concentrations of the solutions of the substance to be examined and of the reference preparation of low-molecular-mass heparins, and calculate the potency of the substance being examined in International Units of anti-factor IIa activity per mL using the usual statistical methods for parallel-line assays.
LABELLING
The label states the number of IU (Units) of anti-factor Xa per unit volume.


