


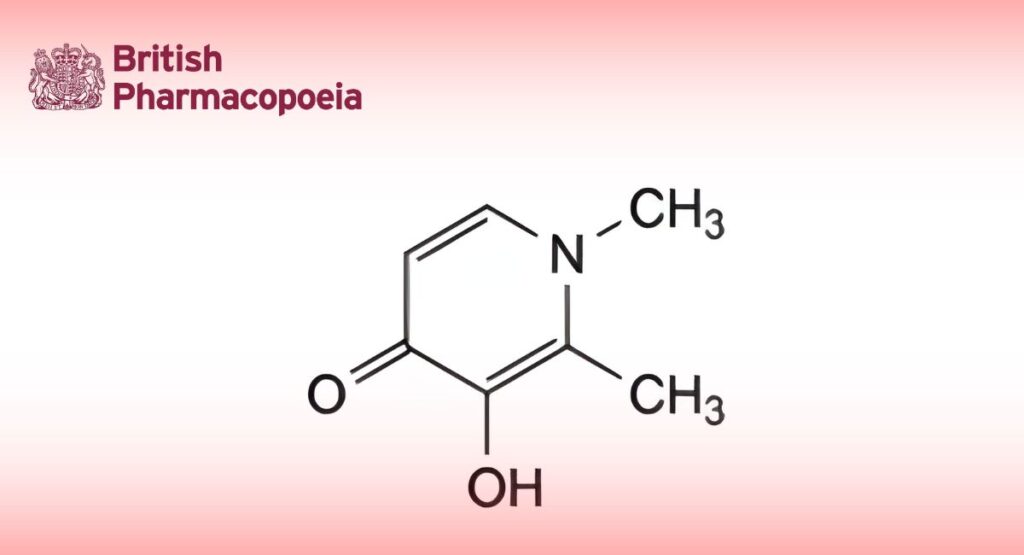
(Ph. Eur. monograph 2236)
C7H9NO2 139.2 30652-11-0
Action and use
Chelating agent (iron).
Preparations
Deferiprone Oral Solution
Deferiprone Tablets
DEFINITION
3-Hydroxy-1,2-dimethylpyridin-4-(1H)-one.
Content
98.0 per cent to 102.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or pinkish-white powder.
Solubility
Sparingly soluble in water, slightly soluble in anhydrous ethanol, practically insoluble in heptane.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparison deferiprone CRS.
TESTS
Related substances
Liquid chromatography (2.2.29). Use only colourless glassware. Protect the solutions from light.
Solution A: Dissolve 2.91 g of sodium edetate R, 4.01 g of sodium octanesulfonate monohydrate R and 6.20 g of
dipotassium hydrogen phosphate R in water for chromatography R and dilute to 2000 mL with the same solvent; adjust to pH 3.0 with phosphoric acid R.
Solution B: Dissolve 0.73 g of sodium edetate R, 1.0 g of sodium octanesulfonate monohydrate R and 1.55 g of
dipotassium hydrogen phosphate R in water for chromatography R and dilute to 2000 mL with the same solvent; adjust to pH 3.0 with phosphoric acid R.
Solvent mixture acetonitrile R, water R (10:90 V/V).
Test solution (a): Dissolve 0.100 g of the substance to be examined in a volume of the mobile phase corresponding to about 2/3 of the final volume and dilute to 100.0 mL with the mobile phase.
Test solution (b): Dissolve 50.0 mg of the substance to be examined in a volume of the solvent mixture corresponding to about 2/3 of the final volume and dilute to 50.0 mL with the solvent mixture. Dilute 5.0 mL of the solution to 200.0 mL with the mobile phase described under Assay.
Reference solution (a) Dissolve 2 mg of maltol R (impurity B) in the mobile phase and dilute to 100.0 mL with the mobile phase. To 2.5 mL of the solution add 10 mL of test solution (a) and dilute to 100.0 mL with the mobile phase.
Reference solution (b) Dilute 1.0 mL of test solution (a) to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 20.0 mL with the mobile phase.
Reference solution (c) Dissolve 50.0 mg of deferiprone CRS in a volume of the solvent mixture corresponding to
about 2/3 of the final volume and dilute to 50.0 mL with the solvent mixture. Dilute 5.0 mL of the solution to 200.0 mL with the mobile phase described under Assay.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: styrene-divinylbenzene copolymer R (5 μm);
— temperature: 30 °C.
Mobile phase acetonitrile R, solution A (10:90 V/V).
Flow rate 1.0 mL/min.
Detection: Spectrophotometer at 280 nm.
Preconditioning of the column: Rinse for 20 min with the mobile phase before each series of injections.
Injection 20 μL of test solution (a) and reference solutions (a) and (b).
Run time 3.5 times the retention time of deferiprone.
Identification of impurities: Use the chromatogram obtained with reference solution (a) to identify the peak due to
impurity B.
Relative retention: With reference to deferiprone (retention time = about 12 min): impurity B = about 0.5.
System suitability: Reference solution (a):
— resolution: minimum 5.0 between the peaks due to impurity B and deferiprone.
Calculation of percentage contents:
— for each impurity, use the concentration of deferiprone in reference solution (b).
Limits:
— impurity B: maximum 0.10 per cent;
— unspecified impurities: for each impurity, maximum 0.05 per cent;
— total: maximum 0.2 per cent;
— reporting threshold: 0.03 per cent.
Water (2.5.32)
Maximum 0.5 per cent, determined on 0.100 g by direct sample introduction.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modifications.
Mobile phase acetonitrile R, solution B (10:90 V/V).
Injection 10 μL of test solution (b) and reference solution (c).
Run time Twice the retention time of deferiprone.
Retention time Deferiprone = about 7.7 min.
Calculate the percentage content of C7H9NO2 taking into account the assigned content of deferiprone CRS.
IMPURITIES
Specified impurities B.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities. It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) A, C.
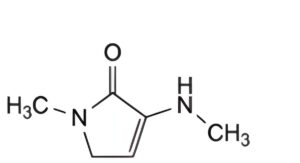
A. 1-methyl-3-(methylamino)-1,5-dihydro-2H-pyrrol-2-one,
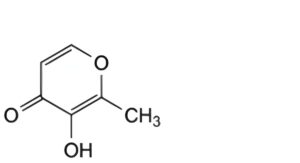
B. 3-hydroxy-2-methyl-4H-pyran-4-one (maltol),
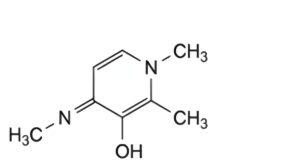
C. 1,2-dimethyl-4-[(Ξ)-methylimino]-1,4-dihydropyridin-3-ol.


