


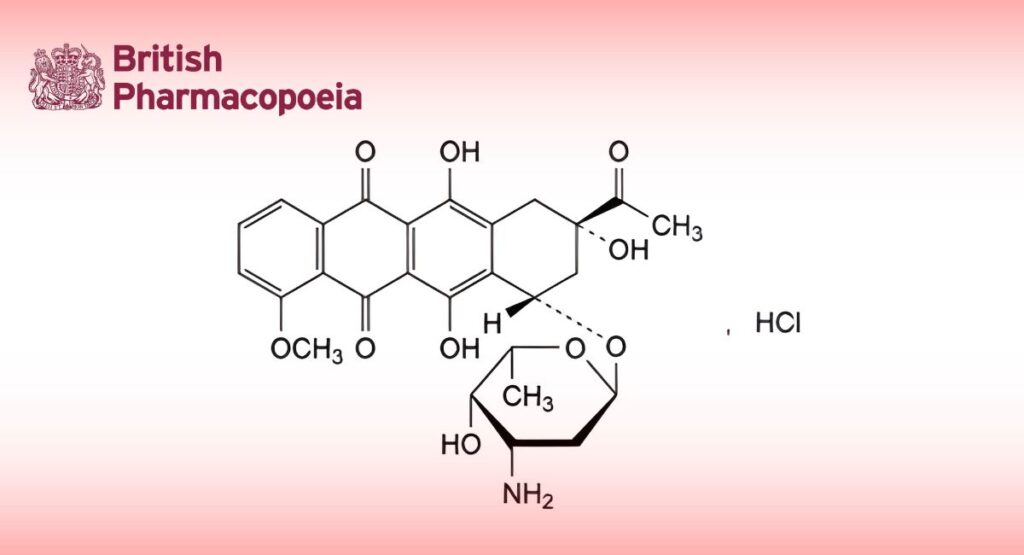
(Ph. Eur. monograph 0662)
C27H30ClNO10 564.0. 23541-50-6
Action and use
Cytostatic; anthracycline antibacterial.
DEFINITION
(8S,10S)-8-Acetyl-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-1-methoxy-7,8,9,10-
tetrahydrotetracene-5,12-dione hydrochloride.
Substance produced by certain strains of Streptomyces coeruleorubidus or Streptomyces peucetius.
Content
95.0 per cent to 102.0 per cent (anhydrous substance).
PRODUCTION
It is produced by methods of manufacture designed to eliminate or minimise the presence of histamine.
CHARACTERS
Appearance
Crystalline, orange-red powder, hygroscopic.
Solubility
Freely soluble in water and in methanol, slightly soluble in ethanol (96 per cent), practically insoluble in acetone.
IDENTIFICATION
A. Infrared absorption spectrophotometry (2.2.24).
Comparison daunorubicin hydrochloride CRS.
B. Dissolve about 10 mg in 0.5 mL of nitric acid R, add 0.5 mL of water R and heat over a flame for 2 min. Allow to cool and add 0.5 mL of silver nitrate solution R1. A white precipitate is formed.
TESTS
pH (2.2.3)
4.5 to 6.5.
Dissolve 50 mg in carbon dioxide-free water R and dilute to 10 mL with the same solvent.
Related substances
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Test solution: Dissolve 50.0 mg of the substance to be examined in the mobile phase and dilute to 50.0 mL with the mobile phase.
Reference solution (a): Dilute 1.0 mL of the test solution to 200.0 mL with the mobile phase.
Reference solution (b): Dissolve the contents of a vial of daunorubicin for system suitability CRS (containing impurities A, B and D) in 1 mL of mobile phase.
Reference solution (c): Dissolve 50.0 mg of daunorubicin hydrochloride CRS in the mobile phase and dilute to 50.0 mL with the mobile phase.
Column:
— size: l = 0.25 m, Ø = 4.0 mm;
— stationary phase: base-deactivated end-capped octadecylsilyl silica gel for chromatography R (5 μm);
— temperature: 25 °C.
Mobile phase Mix 43 volumes of acetonitrile R and 57 volumes of a solution containing 2.25 g/L of phosphoric acid R and 2.88 g/L of sodium laurilsulfate R.
Flow rate 1.0 mL/min.
Detection: Spectrophotometer at 254 nm.
Injection 5 μL of the test solution and reference solutions (a) and (b).
Run time 2.5 times the retention time of daunorubicin.
Identification of impurities: Use the chromatogram supplied with daunorubicin for system suitability CRS and the
chromatogram obtained with reference solution (b) to identify the peaks due to impurities A, B and D.
Relative retention: With reference to daunorubicin (retention time = about 27 min): impurity A = about 0.3;
impurity D = about 0.5; impurity B = about 0.6.
System suitability: Reference solution (b):
— resolution: minimum 2.5 between the peaks due to impurities D and B.
Calculation of percentage contents:
— for each impurity, use the concentration of daunorubicin hydrochloride in reference solution (a).
Limits:
— impurity B: maximum 1.0 per cent;
— impurities A, D: for each impurity, maximum 0.5 per cent;
— any other impurity: for each impurity, maximum 0.5 per cent;
— total (excluding impurities A, B and D): maximum 1.5 per cent;
— reporting threshold: 0.05 per cent.
Butanol (2.4.24, System B)
Maximum 1.0 per cent.
Water (2.5.12)
Maximum 3.0 per cent, determined on 0.100 g.
Bacterial endotoxins (2.6.14)
Less than 4.3 IU/mg, if intended for use in the manufacture of parenteral preparations without a further appropriate procedure for the removal of bacterial endotoxins.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modification.
Injection Test solution and reference solution (c).
Calculate the percentage content of C27H30ClNO10 taking into account the assigned content of daunorubicin
hydrochloride CRS.
STORAGE
In an airtight container, protected from light. If the substance is sterile, the container is also sterile and tamper-evident.
IMPURITIES
Specified impurities A, B, D.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities. It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) C, E, F, G.
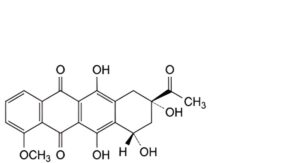
A. (8S,10S)-8-acetyl-6,8,10,11-tetrahydroxy-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (daunorubicin aglycone,daunorubicinone),
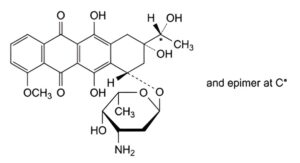
B. (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-8-[(1RS)-1-hydroxyethyl]-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione ((13RS)-daunorubicinol),
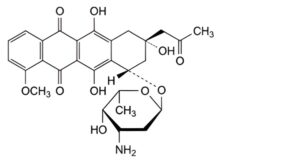
C. (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-1-methoxy-8-(2-
oxopropyl)-7,8,9,10-tetrahydrotetracene-5,12-dione (feudomycin B),
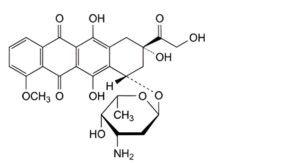
D. (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (doxorubicin),
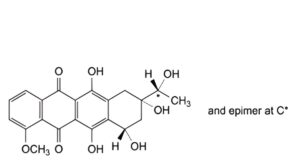
E. (8S,10S)-6,8,10,11-tetrahydroxy-8-[(1RS)-1-hydroxyethyl]-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione
((13RS)-daunorubicinol aglycone, 13-dihydrodaunorubicinone),
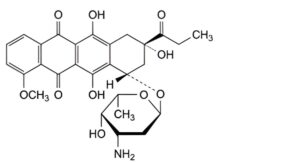
F. (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-6,8,11-trihydroxy-1-methoxy-8-propanoyl-7,8,9,10-tetrahydrotetracene-5,12-dione (14-methyldaunorubicin),
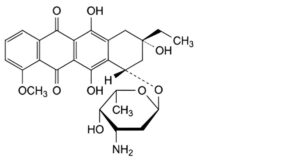
G. (8S,10S)-10-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-8-ethyl-6,8,11-trihydroxy-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (feudomycin A).


