


(Ph. Eur. monograph 2090)
Action and use
Heparinoid; prevention of deep vein thrombosis.
DEFINITION
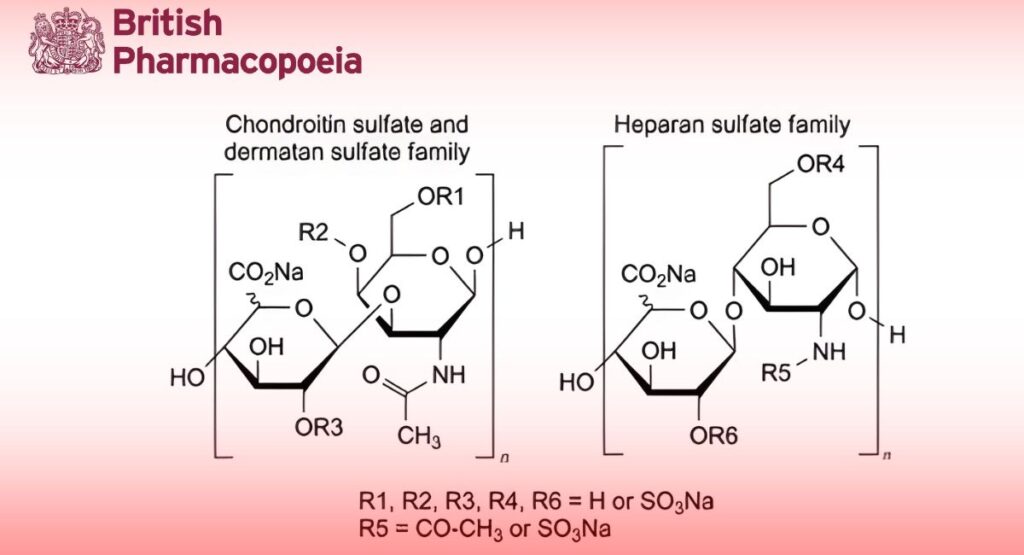
Preparation containing the sodium salts of a mixture of sulfated glycosaminoglycans present in porcine tissues.
Danaparoid sodium is prepared from the intestinal mucosa of pigs. Its major constituents are heparan sulfate and dermatan sulfate. On complete hydrolysis it liberates D-glucosamine, D-galactosamine, D-glucuronic acid, L-iduronic acid, acetic acid and sulfuric acid. It has the characteristic property of enhancing the inactivation of activated factor X (factor Xa) by antithrombin. It has a negligible effect on the inactivation rate of thrombin by antithrombin.
Potency
11.0 to 19.0 anti-factor Xa units per milligram (dried substance).
PRODUCTION
The animals from which danaparoid sodium is derived must fulfil the requirements for the health of animals suitable for human consumption. Danaparoid sodium is prepared using a process that ensures that the relative proportion of active sulfated glycosaminoglycans is consistent. It is produced by methods of manufacturing designed to minimise or eliminate endotoxins and hypotensive substances.
CHARACTERS
Appearance
White or almost white, hygroscopic powder.
Solubility
Freely soluble in water.
IDENTIFICATION
A. The ratio of anti-factor Xa activity to anti-factor IIa activity, determined as described under Assay and
Tests respectively, is not less than 22.
B. Molecular mass distribution (see Tests): the mass-average relative molecular mass ranges between 4000 and 7000.
TESTS
pH (2.2.3)
5.5 to 7.0.
Dissolve 0.5 g of the dried substance to be examined in carbon dioxide-free water R and dilute to 50 mL with the same solvent.
Anti-factor IIa activity
Maximum 0.5 units per milligram (dried substance).
Carry out the test at room temperature. Human thrombin solution R1 must be kept on ice until use.
Test solutions: Prepare 2 independent series of dilutions in geometric progression of the substance to be examined in phosphate buffer solution pH 6.5 R and in the concentration range of 0.0005-0.005 units of anti-factor IIa activity per millilitre.
Reference solutions: Prepare 2 independent series of dilutions in geometric progression of danaparoid sodium CRS in phosphate buffer solution pH 6.5 R and in the concentration range of 0.0005-0.005 units of anti-factor IIa activity per millilitre.
Transfer 50 μL of each solution into the wells of a 96-well microplate. To each well add 50 μL of antithrombin III solution R3 and 50 μL of human thrombin solution R1. Shake the microplate but do not allow bubbles to form. Incubate for 75 min. To each well add 50 μL of chromogenic substrate R4. Shake the microplate. Measure the absorbance at 405 nm (2.2.25) using a suitable reading device, exactly 2 min after the addition of the chromogenic substrate. Measure the absorbance again at 405 nm (2.2.25), exactly 22 min after the addition of the chromogenic substrate. Use ΔA (A22 – A2) for the calculation of the anti-factor IIa activity. Determine the blank amidolytic activity in a similar manner, using phosphate buffer
solution pH 6.5 R as the blank solution (minimum 8 blanks per microplate). Calculate the activity of the substance to be examined in units of anti-factor IIa activity per milligram using a suitable statistical method, for example the parallel-line assay (5.3).
Chondroitin sulfate and dermatan sulfate
Chondroitin sulfate: maximum 8.5 per cent (dried substance); dermatan sulfate: 8.0 per cent to 16.0 per cent (dried substance).
Determine by selective enzymatic degradation.
Test solutions: Dry the substance to be examined at 60 °C over diphosphorus pentoxide R at a pressure of about 670 Pa for 3 h. Dissolve 0.200 g of the dried substance in 10.0 mL of water R. Dilute this solution as necessary to obtain 3 test solutions containing 20 mg/mL, 10 mg/mL and 5 mg/mL of the dried substance to be examined in water R.
Chondroitin sulfate reference solutions: Dry chondroitin sulfate sodium CRS over diphosphorus pentoxide R at room temperature at a pressure of about 670 Pa for 16 h. Prepare solutions containing 1 mg/mL, 2 mg/mL and 3 mg/mL of dried chondroitin sulfate sodium CRS in water R.
Dermatan sulfate reference solutions: Dry dermatan sulfate CRS over diphosphorus pentoxide R at room temperature at a pressure of about 670 Pa for 16 h. Prepare solutions containing 1 mg/mL, 2 mg/mL and 3 mg/mL of dried dermatan sulfate CRS in water R.
Chondroitinase ABC solution: Dissolve chondroitinase ABC R in tris-sodium acetate-sodium chloride buffer solution pH 8.0 R to obtain an activity of 0.5-1.0 units per millilitre.
Chondroitinase AC solution: Dissolve chondroitinase AC R in tris-sodium acetate-sodium chloride buffer solution pH 7.4 R to obtain an activity of 1.0-2.0 units per millilitre.
Procedure:
— Degradation with chondroitinase ABC. Label 2 sets of 10 tubes in triplicate: T1, T2 and T3 for the test solutions; SD1, SD2 and SD3 for the dermatan sulfate reference solutions; SC1, SC2 and SC3 for the chondroitin sulfate reference solutions; and B for the blank (water R). To each tube add 1.25 mL of tris sodium acetate buffer solution pH 8.0 R and 150 μL of the test solutions, dermatan sulfate reference solutions, chondroitin sulfate reference solutions or water R. To each tube in 1 set of tubes add 75 μL of chondroitinase ABC solution. To determine the blank response level, add 75 μL of tris-sodium acetate-sodium chloride buffer solution pH 8.0 R to each tube in the other set of tubes.
Mix the contents of the tubes using a vortex mixer, cover with appropriate stoppers and incubate at 37 °C for at least 24 h.
— Degradation with chondroitinase AC. Label 7 tubes in triplicate: T1, T2 and T3 for the test solutions; SC1, SC2 and SC3 for the chondroitin sulfate reference solutions; and B for the blank (water R). To each tube add 1.25 mL of tris-sodium acetate buffer solution pH 7.4 R and 150 μL of the test solutions,
chondroitin sulfate reference solutions or water R. Add 75 μL of chondroitinase AC solution to each tube. Mix the contents of the tubes using a vortex mixer, cover with appropriate stoppers and incubate at 37 °C for at least 24 h.
After the incubation period mix the contents of the tubes using a vortex mixer and dilute to 12 times with water R. Measure the absorbances (2.2.25) of the diluted solutions at 234 nm against water R using a suitable spectrophotometer.
Calculation: Calculate the mean blank absorbance of each reference solution, i.e. the mean of the absorbances of the reference solutions to which no chondroitinase ABC has been added. Subtract the mean blank absorbance value from the individual absorbance of each reference solution. Calculate linear regression curves for the chondroitin sulfate reference solutions and the dermatan sulfate reference solutions by plotting the blank-corrected absorbances against the concentrations.
Calculate the average percentage content of dermatan sulfate in the test solutions for all tested concentrations using the following expression:
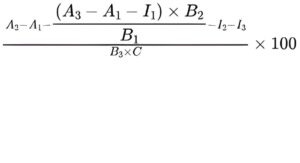
A1 = blank absorbance of the test solution;
A2 = absorbance of the test solution with chondroitinase ABC;
A3 = absorbance of the test solution with chondroitinase AC;
B1 = gradient of the curve obtained with the chondroitin sulfate reference solutions with chondroitinase AC;
B2 = gradient of the curve obtained with the chondroitin sulfate reference solutions with chondroitinase ABC;
B3 = gradient of the curve obtained with the dermatan sulfate reference solutions with chondroitinase ABC;
C = concentration of the test solution, in milligrams per millilitre;
I1 = y-intercept of the curve obtained with the chondroitin sulfate reference solutions with chondroitinase AC;
I2 = y-intercept of the curve obtained with the chondroitin sulfate reference solutions with chondroitinase ABC;
I3 = y-intercept of the curve obtained with the dermatan sulfate reference solutions with chondroitinase ABC.
Calculate the average percentage content of chondroitin sulfate in the test solutions for all tested concentrations using the following expression:
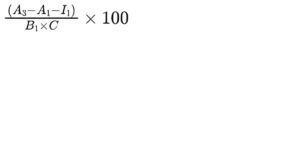
Molecular mass distribution
Size-exclusion chromatography (2.2.30).
Test solution: Dissolve 10 mg of the substance to be examined in 2 mL of the mobile phase.
Reference solution: Dissolve 10 mg of danaparoid sodium CRS in 2 mL of the mobile phase.
Column:
— size: l = 0.60 m, Ø = 7.5 mm;
— stationary phase: hydrophilic silica gel for chromatography R (10 μm) with a fractionation range for proteins with a relative molecular mass of approximately 5000 to 100 000;
— temperature: 30 °C.
Mobile phase: 28.4 g/L solution of anhydrous sodium sulfate R adjusted to pH 5.0 with dilute sulfuric acid R.
Flow rate: 0.9 mL/min ± 2 per cent.
Detection: Spectrophotometer at 210 nm.
Injection: 100 μL.
Run time: For a period of time ensuring complete elution of sample and solvent peaks (about 40 min).
System suitability: Inject the reference solution twice; the difference between the retention times corresponding to the maxima of the peaks is not more than 5 s.
Calibration: Calibration is achieved by taking the relevant part of the chromatogram obtained with the reference solution, i.e. excluding the sharp peak at the end of the chromatogram, and matching the chromatogram obtained with the test solution with the calibration table obtained with the reference solution. From the calibration curve obtained, determine the molecular mass distribution of the sample. A calibration table is supplied with danaparoid sodium CRS.
Limits:
— chains with a relative molecular mass less than 2000: maximum 13 per cent;
— chains with a relative molecular mass less than 4000: maximum 39 per cent;
— chains with a relative molecular mass between 4000 and 8000: minimum 50 per cent;
— chains with a relative molecular mass higher than 8000: maximum 19 per cent;
— chains with a relative molecular mass higher than 10 000: maximum 11 per cent.
Nitrogen (2.5.9)
2.4 per cent to 3.0 per cent (dried substance).
Nucleic acids
Maximum 0.5 per cent (dried substance).
Test solution: Weigh about 50 mg of the dried substance to be examined into a centrifuge tube and dissolve in 200 μL of water R.
Reference solution: Dissolve about 50 mg of ribonucleic acid CRS in 5 mL of 0.1 M sodium hydroxide and dilute to 20.0 mL with water R. Transfer 200 μL of the solution into a centrifuge tube.
Add 4.0 mL of a 50 g/L solution of trichloroacetic acid R to each tube and mix. Place all tubes in boiling water for 30 min.
Allow to cool to room temperature. Add again 4.0 mL of a 50 g/L solution of trichloroacetic acid R to each tube and mix. If any of the test solutions is not clear, sonicate all the tubes in an ultrasonic bath for 10 min and centrifuge at 1500 g for 15 min. Dilute 1.0 mL of the clear supernatant to 4.0 mL with water R. Measure the absorbances of the diluted reference and test solutions at 265 nm (2.2.25) against a blank solution prepared in the same manner, and calculate the percentage nucleic acid content of the sample.
Total protein (2.5.33, Method 2)
Maximum 0.5 per cent.
Dissolve the substance to be examined in distilled water R. Use bovine albumin R1 as the reference substance.
Adjust the concentration of the diluted phosphomolybdotungstic reagent so that the pH in the reaction mixture is between 10.0 and 10.5.
Sodium
9.0 per cent to 11.0 per cent (dried substance).
Atomic absorption spectrometry (2.2.23, Method I).
Test solution: Dissolve 0.125 g of the substance to be examined in 100.0 mL of a 1.27 mg/mL solution of caesium chloride R in 0.1 M hydrochloric acid.
Reference solutions: Prepare reference solutions containing 50 ppm, 100 ppm and 150 ppm of Na by diluting sodium standard solution (1000 ppm Na) R with a 1.27 mg/mL solution of caesium chloride R in 0.1 M hydrochloric acid.
Source: Sodium hollow-cathode lamp.
Wavelength: 330.3 nm.
Atomisation device: Air-acetylene flame.
Loss on drying (2.2.32)
Maximum 5.0 per cent, determined on 0.500 g by drying in an oven at 60 °C over diphosphorus pentoxide R at a pressure of 670 Pa for 3 h.
Bacterial endotoxins (2.6.14)
Less than 0.02 IU per unit of anti-factor Xa activity, if intended for use in the manufacture of parenteral preparations without a further appropriate procedure for the removal of bacterial endotoxins.
ASSAY
Carry out the assay at room temperature.
The anticoagulant activity of danaparoid sodium is determined in vitro by an assay which determines its ability to accelerate the inhibition of factor Xa by antithrombin III (anti-factor Xa assay).
Test solutions: Prepare 2 independent series of dilutions in geometric progression of the substance to be examined in tris(hydroxymethyl)aminomethane EDTA buffer solution pH 8.4 R and in the concentration range of 0.1-0.32 units of anti- factor Xa activity per millilitre.
Reference solutions: Prepare 2 independent series of dilutions in geometric progression of danaparoid sodium CRS in tris(hydroxymethyl)aminomethane EDTA buffer solution pH 8.4 R and in the concentration range of 0.08-0.35 units of anti- factor Xa activity per millilitre.
Transfer 40 μL of each solution into the wells of a 96-well microplate. Add 40 μL of antithrombin III solution R4 to each well and shake the microplate but do not allow bubbles to form. Add 40 μL of bovine factor Xa solution R1 to each well. Exactly 2 min after the addition of the factor Xa solution, add 80 μL of chromogenic substrate R5. Measure the absorbance at 405 nm (2.2.25) using a suitable reading device, exactly 4 min after the addition of the factor Xa solution. Measure the absorbance again at 405 nm (2.2.25), exactly 14 min after the addition of the factor Xa solution. Use ΔA (A14 – A4) for the calculation of the anti-factor Xa activity. Determine the blank amidolytic activity in the same manner, using
tris(hydroxymethyl)aminomethane EDTA buffer solution pH 8.4 R as the blank (minimum 8 blanks per microplate).
Calculate the potency of the substance to be examined in units of anti-factor Xa activity per milligram using a suitable statistical method, for example the parallel-line assay (5.3).
STORAGE
In an airtight container. If the substance is sterile, the container is also sterile and tamper-evident.
LABELLING
The label states the number of units of anti-factor Xa activity per milligram.


