


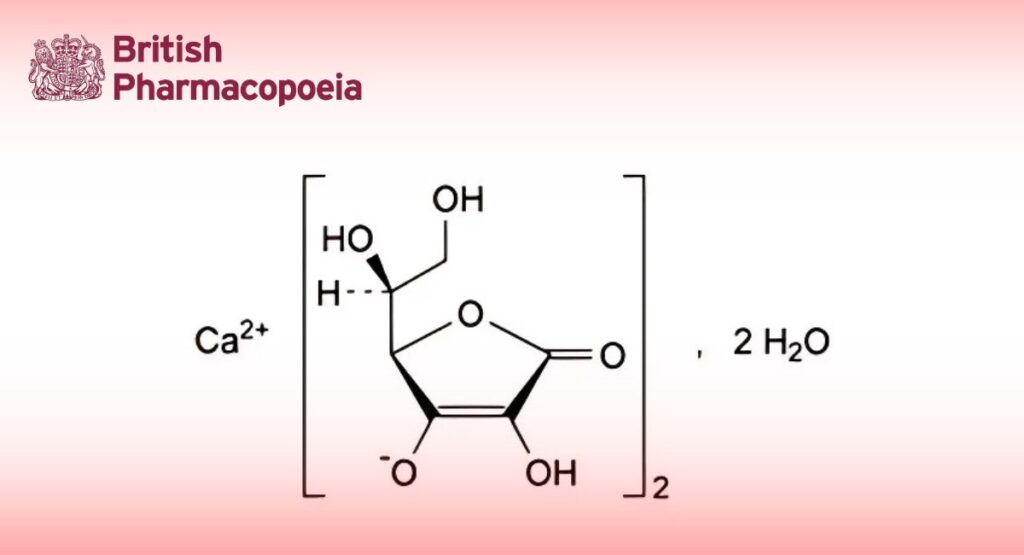
(Ph. Eur. monograph 1182)
C12H14CaO12,2H2O 426.3 5743-28-2
Action and use
Excipient.
DEFINITION
Calcium di[(2R)-2-[(1S)-1,2-dihydroxyethyl]-4-hydroxy-5-oxo-2,5-dihydrofuran-3-olate] dihydrate.
Content
99.0 per cent to 100.5 per cent (dried substance).
CHARACTERS
Appearance
White or slightly yellowish, crystalline powder.
Solubility
Freely soluble in water, practically insoluble in ethanol (96 per cent) and in heptane.
IDENTIFICATION
First identification: A, B, E.
Second identification: A, C, D, E.
A. Specific optical rotation (see Tests).
B. Infrared absorption spectrophotometry (2.2.24).
Comparison: calcium ascorbate dihydrate CRS.
C. Dilute 1 mL of solution S (see Tests) to 10 mL with water R. To 2 mL of the solution add 0.2 mL of a 100 g/L solution of ferrous sulfate R. A deep violet colour develops.
D. To 1 mL of solution S add 0.2 mL of dilute nitric acid R and 0.2 mL of silver nitrate solution R2. A grey precipitate is formed.
E. The substance gives reaction (b) of calcium (2.3.1).
TESTS
Solution S
Dissolve 5.00 g in carbon dioxide-free water R and dilute to 50.0 mL with the same solvent.
Appearance of solution
Solution S is clear (2.2.1) and not more intensely coloured than reference solution Y6 (2.2.2, Method II).
Examine the colour of the solution immediately after preparation.
pH (2.2.3)
6.8 to 7.4 for solution S.
Specific optical rotation (2.2.7)
+ 95 to + 97 (dried substance), determined using freshly prepared solution S.
Related substances
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Solution A A 17 g/L solution of potassium dihydrogen phosphate R.
Phosphate buffer solution: Dissolve 6.8 g of potassium dihydrogen phosphate R in water for chromatography R and dilute to about 200 mL with the same solvent. Filter through a membrane filter (nominal pore size 0.45 μm) and dilute to 1000 mL with water for chromatography R.
Test solution: Dissolve 0.500 g of the substance to be examined in 2.5 mL of water R and sonicate for 4 min.
After complete dissolution, add 2.5 mL of solution A and dilute to 10.0 mL with acetonitrile R. Centrifuge for 5 min at 1500-2000 g. Inject the clear supernatant.
Reference solution (a): Dissolve 10.0 mg of ascorbic acid impurity C CRS in the mobile phase and dilute to 5.0 mL with the mobile phase.
Reference solution (b): Dissolve 5.0 mg of ascorbic acid impurity D CRS and 5.0 mg of ascorbic acid CRS in the mobile phase and dilute to 100.0 mL with the mobile phase.
Reference solution (c): Dilute 1 mL of the test solution to 200 mL with the mobile phase. Mix 1 mL of this solution and 1 mL of reference solution (a).
Reference solution (d): Dilute 2.5 mL of reference solution (a) to 100.0 mL with the mobile phase.
Column:
— size: l = 0.25 m, Ø = 4.6 mm;
— stationary phase: aminopropylsilyl silica gel for chromatography R (5 μm);
— temperature: 45 °C.
Mobile phase: Phosphate buffer solution, acetonitrile R1 (25:75 V/V).
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 210 nm.
Injection: 20 μL of the test solution and reference solutions (b), (c) and (d).
Run time: 4.5 times the retention time of ascorbic acid.
Identification of impurities: Use the chromatogram obtained with reference solution (d) to identify the peak due to impurity C; use the chromatogram obtained with reference solution (b) to identify the peak due to impurity D.
Relative retention: With reference to ascorbic acid (retention time = about 9 min): impurity D = about 0.5; impurity C = about 1.45.
System suitability:
— resolution: minimum 3.0 between the peaks due to ascorbic acid and impurity C in the chromatogram obtained with reference solution (c);
— signal-to-noise ratio: minimum 20 for the peak due to impurity C in the chromatogram obtained with reference solution (d).
Calculation of percentage contents:
— for impurity C, use the concentration of impurity C in reference solution (d);
— for impurity D, use the concentration of impurity D in reference solution (b);
— for impurities other than C and D, use the concentration of ascorbic acid in reference solution (b).
Limits:
— impurities C, D: for each impurity, maximum 0.15 per cent;
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— sum of impurities other than C and D: maximum 0.2 per cent;
— reporting threshold: 0.05 per cent.
Fluorides
Maximum 10 ppm.
Potentiometry (2.2.36, Method I).
Test solution: In a 50 mL volumetric flask, dissolve 1.000 g in a 10.3 g/L solution of hydrochloric acid R, add 5.0 mL of fluoride standard solution (1 ppm F) R and dilute to 50.0 mL with a 10.3 g/L solution of hydrochloric acid R. To 20.0 mL of the solution add 20.0 mL of total-ionic-strength-adjustment buffer R and 3 mL of an 82 g/L solution of anhydrous sodium acetate R. Adjust to pH 5.2 with ammonia R and dilute to 50.0 mL with distilled water R.
Reference solutions: To 0.25 mL, 0.5 mL, 1.0 mL, 2.0 mL and 5.0 mL of fluoride standard solution (10 ppm F) R add 20.0 mL of total-ionic-strength-adjustment buffer R and dilute to 50.0 mL with distilled water R.
Indicator electrode: Fluoride selective.
Reference electrode: Silver-silver chloride.
Take into account the addition of fluoride to the test solution for the calculation.
Iron
Maximum 2 ppm.
Atomic absorption spectrometry (2.2.23).
Test solution: Dissolve 5.0 g in a 9.7 g/L solution of nitric acid R and dilute to 25.0 mL with the same acid solution.
Reference solutions: Prepare the reference solutions using iron standard solution (10 ppm Fe) R, diluting with a 9.7 g/L solution of nitric acid R.
Source Iron hollow-cathode lamp.
Wavelength 248.3 nm.
Atomisation device Air-acetylene flame.
Loss on drying (2.2.32)
Maximum 0.1 per cent, determined on 1.000 g by drying in an oven at 105 °C for 2 h.
ASSAY
Dissolve 80.0 mg in a mixture of 10 mL of dilute sulfuric acid R and 80 mL of carbon dioxide-free water R. Add 1 mL of starch solution R. Titrate with 0.05 M iodine until a persistent violet-blue colour is obtained.
1 mL of 0.05 M iodine is equivalent to 10.66 mg of C12H14CaO12,2H2O.
STORAGE
In a non-metallic container, protected from light.
IMPURITIES
Specified impurities C, D.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) A, E, F.
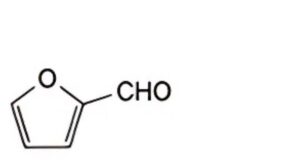
A. furan-2-carbaldehyde (furfural),
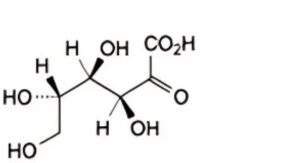
C. L-xylo-hex-2-ulosonic acid (L-sorbosonic acid),
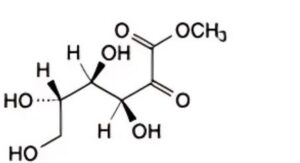
D. methyl L-xylo-hex-2-ulosonate (methyl L-sorbosonate),
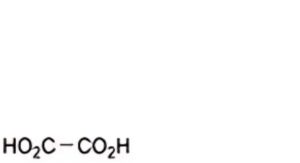
E. oxalic acid,
F. (5R)-5-[(1R)-1,2-dihydroxyethyl]-3,4-dihydroxyfuran-2(5H)-one.


