(Ph. Eur. monograph 1077)
C60H86N16O13 1239 57982-77-1
Action and use
Gonadotrophin releasing hormone (gonadorelin) analogue; treatment of prostate cancer.
DEFINITION
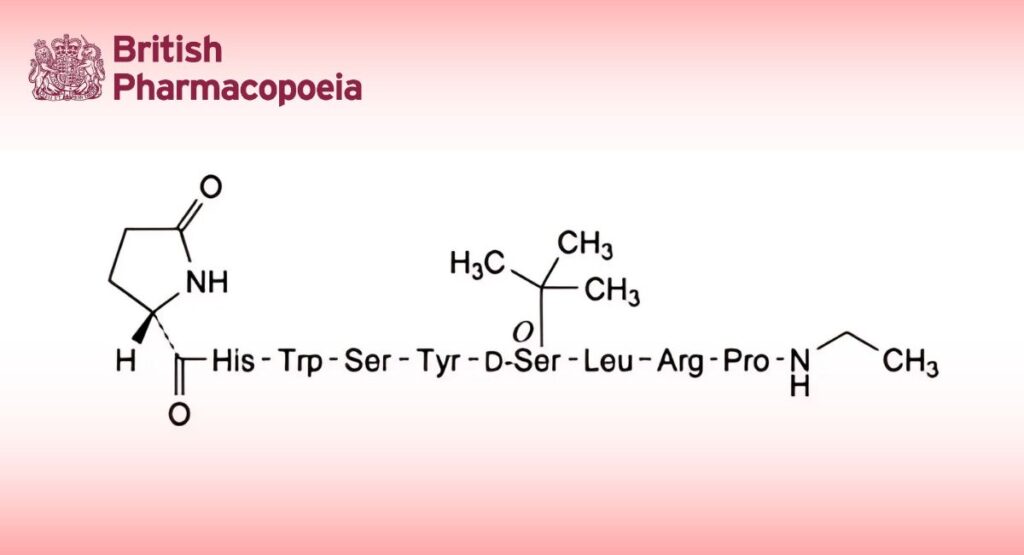
5-Oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-O-(1,1-dimethylethyl)-D-seryl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide.
Synthetic nonapeptide analogue of human gonadotrophin-releasing hormone GnRH with agonistic activity to gonadorelin. It is obtained by chemical synthesis and is available as an acetate.
Content
95.0 per cent to 102.0 per cent (anhydrous, acetic acid-free substance).
CHARACTERS
Appearance
White or slightly yellowish powder, hygroscopic.
Solubility
Sparingly soluble in water and in dilute acids.
IDENTIFICATION
Carry out either tests A and B or tests A and C.
A. Examine the chromatograms obtained in the assay.
Results: The principal peak in the chromatogram obtained with the test solution is similar in retention time and size to the principal peak in the chromatogram obtained with reference solution (b).
B. Nuclear magnetic resonance spectrometry (2.2.64).
Preparation: 4 mg/mL solution in a mixture of 20 volumes of deuterated acetic acid R and 80 volumes of deuterium oxide R.
Comparison: Dissolve the contents of a vial of buserelin for NMR identification CRS in a mixture of 20 volumes of deuterated acetic acid R and 80 volumes of deuterium oxide R to obtain a concentration of 4 mg/mL.
Operating conditions:
— field strength: minimum 300 MHz;
— temperature: 27 °C.
Results: Examine the H NMR spectrum from 0 to 9 ppm. The H NMR spectrum obtained is qualitatively similar to the HNMR spectrum obtained with buserelin for NMR identification CRS.
C. Amino acid analysis (2.2.56). Method 1 for hydrolysis and method 1 for analysis are suitable.
Express the content of each amino acid in moles. Calculate the relative proportions of the amino acids, taking 1/6 of the sum of the number of moles of glutamic acid, histidine, tyrosine, leucine, arginine and proline as equal to 1. The values fall within the following limits: serine 1.4 to 2.0; proline 0.8 to 1.2; glutamic acid 0.9 to 1.1; leucine 0.9 to 1.1; tyrosine 0.9 to 1.1; histidine 0.9 to 1.1; arginine 0.9 to 1.1. Not more than traces of other amino acids are present.
TESTS
Appearance of solution
A 10 g/L solution is clear (2.2.1) and not more intensely coloured than reference solution Y7 (2.2.2, Method II).
Specific optical rotation (2.2.7)
-58 to -49 (anhydrous, acetic acid-free substance), determined on a 10 g/L solution.
Specific absorbance (2.2.25)
49 to 56, measured at the absorption maximum at 278 nm (anhydrous, acetic acid-free substance).
Dissolve 10.0 mg in 100.0 mL of 0.01 M hydrochloric acid.
Related substances
Liquid chromatography (2.2.29).
Test solution: Dissolve 5.0 mg of the substance to be examined in 5.0 mL of the mobile phase.
Reference solution (a): Dissolve the contents of a vial of D-His-buserelin CRS (impurity A) in the mobile phase. Dilute an appropriate volume of this solution in the mobile phase to obtain a final concentration of 1 mg/mL. Add 1.0 mL of the test solution to 1.0 mL of this solution.
Reference solution (b): Dissolve the contents of a vial of buserelin CRS in the mobile phase. Dilute an appropriate volume of this solution in the mobile phase to obtain a final concentration of 1.0 mg/mL.
Reference solution (c): Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase.
Reference solution (d): Dissolve the contents of a vial of buserelin for peak identification CRS (containing impurities F and G) in the mobile phase. Dilute an appropriate volume of this solution in the mobile phase to obtain a final concentration of 1 mg/mL.
Column:
— size: l = 0.25 m, Ø = 4 mm;
— stationary phase: octadecylsilyl silica gel for chromatography R (5 μm);
— temperature: 42 °C.
Mobile phase: Mix 200 mL of acetonitrile R and 700 mL of an 11.2 g/L solution of phosphoric acid R previously adjusted to pH 2.5 with triethylamine R.
Flow rate: 0.8 mL/min.
Detection: Spectrophotometer at 220 nm.
Injection: 10 μL of the test solution and reference solutions (a), (c) and (d).
Run time: 60 min.
Identification of impurities: Use the chromatogram obtained with reference solution (a) to identify the peak due to impurity A and the chromatogram obtained with reference solution (d) to identify the peaks due to impurities F and G.
Relative retention: With reference to buserelin (retention time = about 23 min): impurity F = about 0.83; impurity A = about 0.91; impurity G = about 1.26.
System suitability: Reference solution (a):
— resolution: minimum 1.5 between the peaks due to impurity A and buserelin.
Calculation of percentage contents:
— for each impurity, use the concentration of buserelin in reference solution (c).
Limits:
— impurity A: maximum 2.5 per cent;
— impurity F: maximum 1.0 per cent;
— impurity G: maximum 1.0 per cent;
— unspecified impurities: for each impurity, maximum 0.5 per cent;
— total: maximum 4.0 per cent;
— reporting threshold: 0.1 per cent.
Acetic acid (2.5.34)
3.0 per cent to 7.0 per cent.
Test solution Dissolve 20.0 mg of the substance to be examined in a mixture of 5 volumes of mobile phase B and 95 volumes of mobile phase A and dilute to 10.0 mL with the same mixture of solvents.
Water (2.5.12)
Maximum 4.0 per cent, determined on 80.0 mg.
Bacterial endotoxins (2.6.14)
Less than 55.5 IU/mg, if intended for use in the manufacture of parenteral preparations without a further appropriate procedure for the removal of bacterial endotoxins.
ASSAY
Liquid chromatography (2.2.29) as described in the test for related substances with the following modification.
Injection: Test solution and reference solution (b).
Calculate the percentage content of buserelin (C60H86N16O13) taking into account the assigned content of C60H86N16O13 in buserelin CRS.
STORAGE
In an airtight container, protected from light, at a temperature of 2 °C to 8 °C. If the substance is sterile, the container is also sterile and tamper-evident.
LABELLING
The label states:
— the mass of peptide in the container;
— where applicable, that the substance is suitable for use in the manufacture of parenteral preparations.
IMPURITIES
Specified impurities: A, F, G.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B, C, D, E.
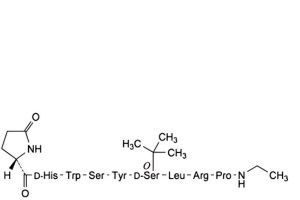
A. [2-D-histidine]buserelin,
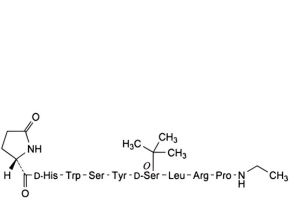
B. [4-D-serine]buserelin,
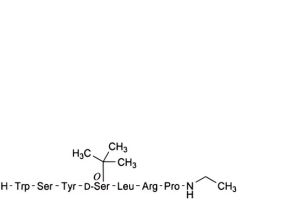
C. buserelin-(3-9)-peptide,
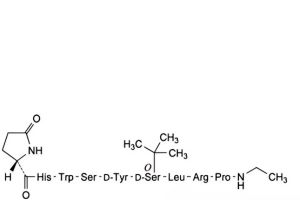
D. [5-D-tyrosine]buserelin,
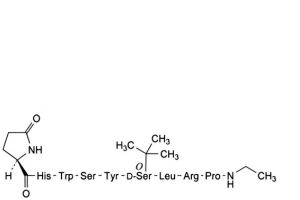
E. [1-(5-oxo-D-proline)]buserelin,
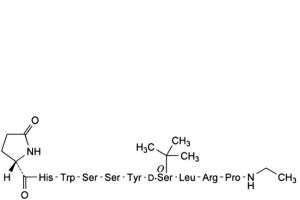
F. endo-3a-serine-buserelin,
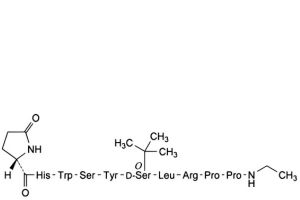
G. endo-8a-proline-buserelin.