


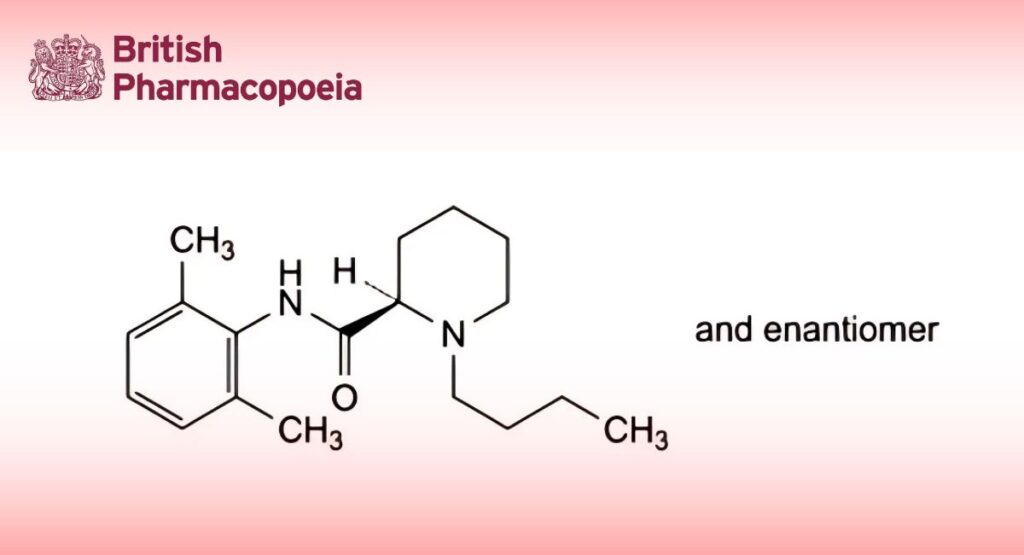
(Ph. Eur. monograph 2761)
C18H28N2O 288.4 38396-39-3
DEFINITION
(2RS)-1-Butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide.
Content
99.0 per cent to 101.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white, crystalline powder, crystals or granules.
Solubility
Practically insoluble in water, freely soluble in ethanol (96 per cent), slightly soluble in heptane.
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparison: bupivacaine CRS.
TESTS
Appearance of solution
The solution is clear (2.2.1) and not more intensely coloured than reference solution BY7 (2.2.2, Method II).
Dissolve 1.0 g in ethyl acetate R and dilute to 10 mL with the same solvent.
Impurity F
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Solvent mixture methanol R, water R (10:90 V/V).
Test solution: Dissolve 0.500 g of the substance to be examined in methanol R and dilute to 5.0 mL with the same solvent.
Reference solution: Dissolve 25.0 mg of bupivacaine impurity F CRS in methanol R and dilute to 20.0 mL with the same solvent. Dilute 2.0 mL of the solution to 100.0 mL with the solvent mixture. Dilute 1.0 mL of this solution to 100.0 mL with the solvent mixture.
Column:
— size: l = 0.05 m, Ø = 4.6 mm;
— stationary phase: end-capped ethylene-bridged octadecylsilyl silica gel for chromatography (hybrid material) R (3.5 μm).
Mobile phase:
— mobile phase A: methanol R2, 0.1 per cent V/V solution of phosphoric acid R (10:90 V/V);
— mobile phase B: methanol R2;
| Time
(min) |
Mobile phase A
(per cent V/V) |
Mobile phase B
(per cent V/V) |
| 0 – 10 | 100 | 0 |
| 10 – 15 | 100 → 5 | 0 → 95 |
Flow rate: 1.5 mL/min.
Detection: Spectrophotometer at 210 nm.
Injection: 5 μL.
Retention time: Impurity F = about 2.5 min.
System suitability: Reference solution:
— signal-to-noise ratio: minimum 20 for the peak due to impurity F.
Calculation of content:
— use the concentration of impurity F in the reference solution.
Limit:
— impurity F: maximum 2.5 ppm.
Related substances
Liquid chromatography (2.2.29).
Buffer solution 0.79 g/L solution of ammonium hydrogen carbonate R adjusted to pH 8.3 with concentrated ammonia R.
Test solution: Dissolve 80.0 mg of the substance to be examined in acetonitrile R and dilute to 100.0 mL with the same solvent.
Reference solution (a): Dilute 1.0 mL of the test solution to 100.0 mL with acetonitrile R. Dilute 1.0 mL of this solution to 10.0 mL with the same solvent.
Reference solution (b): Dissolve the contents of a vial of bupivacaine impurity D CRS in 1 mL of the test solution.
Column:
— size: l = 0.15 m, Ø = 4.6 mm;
— stationary phase: end-capped extra-dense bonded octylsilyl silica gel for chromatography R (5 μm);
— temperature: 22 °C.
Mobile phase:
— mobile phase A: acetonitrile for chromatography R, buffer solution (10:90 V/V);
— mobile phase B: buffer solution, acetonitrile for chromatography R (10:90 V/V);
| Time
(min) |
Mobile phase A
(per cent V/V) |
Mobile phase B
(per cent V/V) |
| 0 – 10 | 100 | 0 |
| 10 – 15 | 100 → 5 | 0 → 95 |
Flow rate: 1.0 mL/min.
Detection: Spectrophotometer at 225 nm.
Autosampler: Set at 15 °C.
Injection: 20 μL.
Identification of impurities: Use the chromatogram obtained with reference solution (b) to identify the peak due to impurity D.
Relative retention: With reference to bupivacaine (retention time = about 24 min): impurity D = about 0.97.
System suitability: Reference solution (b):
— peak-to-valley ratio: minimum 1.5, where Hp = height above the baseline of the peak due to impurity D and
Hv = height above the baseline of the lowest point of the curve separating this peak from the peak due to bupivacaine.
Calculation of percentage contents:
— for each impurity, use the concentration of bupivacaine in reference solution (a).
Limits:
— unspecified impurities: for each impurity, maximum 0.10 per cent;
— total: maximum 1.0 per cent;
— reporting threshold: 0.05 per cent.
Water (2.5.12)
Maximum 0.4 per cent, determined on 0.500 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.220 g in 40 mL of glacial acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point
potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 28.84 mg of C18H28N2O.
IMPURITIES
Specified impurities F.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) A, B, C, D, E, G.
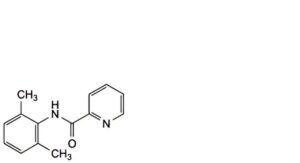
A. N-(2,6-dimethylphenyl)pyridine-2-carboxamide,
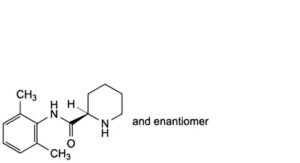
B. (2RS)-N-(2,6-dimethylphenyl)piperidine-2-carboxamide,
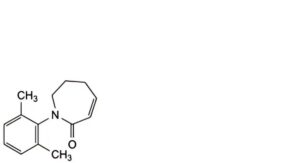
C. 1-(2,6-dimethylphenyl)-1,5,6,7-tetrahydro-2H-azepin-2-one,
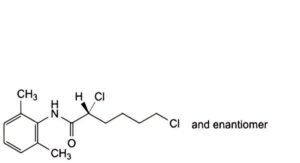
D. (2RS)-2,6-dichloro-N-(2,6-dimethylphenyl)hexanamide,
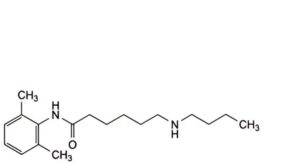
E. 6-(butylamino)-N-(2,6-dimethylphenyl)hexanamide,

F. 2,6-dimethylaniline,

G. (2RS)-1-butyl-N-(2,4-dimethylphenyl)piperidine-2-carboxamide.


