


(Ph. Eur. monograph 0653)
C10H12ClNO2 213.7 1134-47-0
Action and use
Skeletal muscle relaxant.
Preparations
Baclofen Oral Solution
Baclofen Tablets
DEFINITION
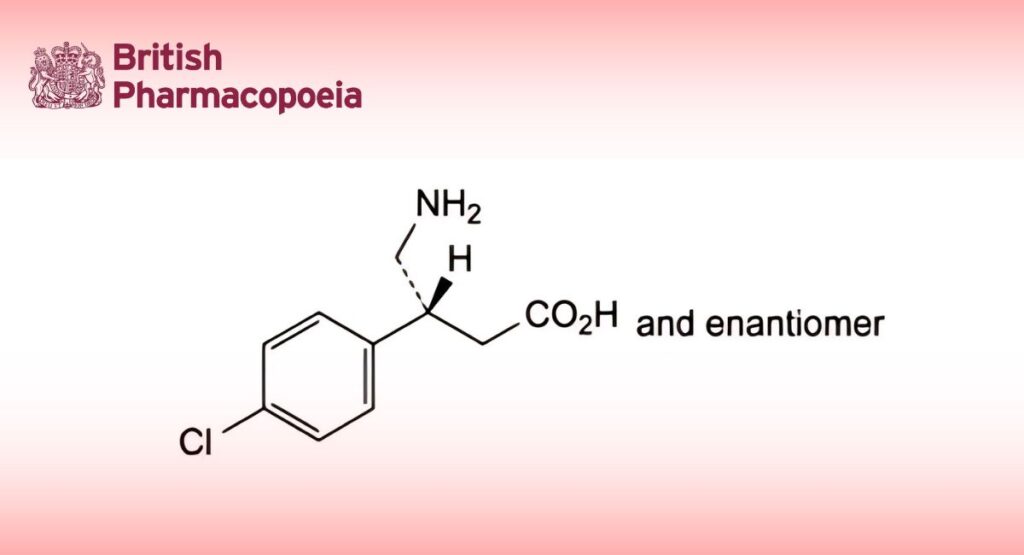
(3RS)-4-Amino-3-(4-chlorophenyl)butanoic acid.
Content
98.0 per cent to 101.0 per cent (anhydrous substance).
CHARACTERS
Appearance
White or almost white powder.
Solubility
Slightly soluble in water, very slightly soluble in ethanol (96 per cent), practically insoluble in acetone. It dissolves in dilute mineral acids and in dilute solutions of alkali hydroxides.
It shows polymorphism (5.9).
IDENTIFICATION
First identification: B.
Second identification: A, C.
A. Ultraviolet and visible absorption spectrophotometry (2.2.25).
Test solution: Dissolve 70 mg in water R and dilute to 100.0 mL with the same solvent.
Spectral range: 220-320 nm.
Absorption maxima: At 259 nm, 266 nm and 275 nm.
Resolution (2.2.25): minimum 1.5 for the absorbance ratio.
Specific absorbance at the absorption maxima:
— at 259 nm: 9.8 to 10.8;
— at 266 nm: 11.5 to 12.7;
— at 275 nm: 8.4 to 9.3.
B. Infrared absorption spectrophotometry (2.2.24).
Preparation: Discs prepared using 3 mg of substance and 300 mg of potassium bromide R.
Comparison baclofen CRS.
If the spectra obtained in the solid state show differences, dissolve 0.1 g of each of the substances separately in 1 mL of dilute sodium hydroxide solution R and add 10 mL of ethanol (96 per cent) R and 1 mL of dilute acetic acid R. Allow to stand for 1 h. Filter, wash the precipitate with ethanol (96 per cent) R and dry in vacuo. Prepare new discs and record the spectra.
C. Thin-layer chromatography (2.2.27).
Test solution: Dissolve 10 mg of the substance to be examined in the mobile phase and dilute to 10 mL with the mobile phase.
Reference solution: Dissolve 10 mg of baclofen CRS in the mobile phase and dilute to 10 mL with the mobile phase.
Plate: TLC silica gel G plate R.
Mobile phase anhydrous formic acid R, water R, methanol R, chloroform R, ethyl acetate R (5:5:20:30:40 V/V/V/V/V).
Application: 5 μL.
Development: Over a path of 12 cm.
Drying: Allow the solvents to evaporate.
Detection Spray with ninhydrin solution R3 until the plate is slightly wet. Place in an oven maintained at 100 °C for 10 min. Examine in daylight.
Results: The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with the reference solution.
TESTS
Appearance of solution
The solution is not more opalescent than reference suspension II (2.2.1) and not more intensely coloured than reference solution BY5 (2.2.2, Method II).
Dissolve 0.50 g in 1 M sodium hydroxide and dilute to 25 mL with the same solvent.
Related substances
Liquid chromatography (2.2.29).
Test solution: Dissolve 25.0 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a): Dissolve 25.0 mg of baclofen impurity A CRS in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (b): Dilute 1.0 mL of reference solution (a) to 100.0 mL with the mobile phase.
Reference solution (c): Dilute 2.0 mL of the test solution to 100.0 mL with the mobile phase.
Reference solution (d): Dilute 2.0 mL of the test solution and 2.0 mL of reference solution (a) to 100.0 mL with the mobile phase.
Column:
— size: l = 0.25 m, Ø = 4.0 mm;
— stationary phase: octadecylsilyl silica gel for chromatography R (10 μm).
Mobile phase: Dissolve 1.822 g of sodium hexanesulfonate R in 1 L of a mixture of 560 volumes of water R, 440 volumes of methanol R and 5 volumes of glacial acetic acid R.
Flow rate: 2.0 mL/min.
Detection: Spectrophotometer at 266 nm.
Injection: 20 μL of the test solution and reference solutions (b), (c) and (d).
Run time: 5 times the retention time of baclofen.
System suitability: Reference solution (d):
— resolution: minimum 2.0 between the peaks due to baclofen and impurity A.
Limits:
— impurity A: not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (1.0 per cent);
— total: not more than the area of the principal peak in the chromatogram obtained with reference solution (c) (2.0 percent).
Water (2.5.12)
Maximum 1.0 per cent, determined on 1.000 g.
Sulfated ash (2.4.14)
Maximum 0.1 per cent, determined on 1.0 g.
ASSAY
Dissolve 0.1500 g in 50 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 21.37 mg of C10H12ClNO2.
IMPURITIES
Specified impurities A.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use) B.
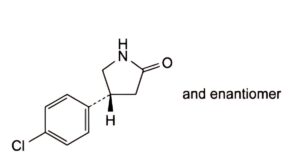
A. (4RS)-4-(4-chlorophenyl)pyrrolidin-2-one,
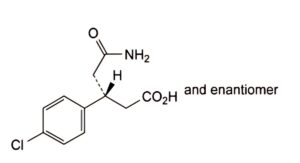
B. (3RS)-5-amino-3-(4-chlorophenyl)-5-oxopentanoic acid.


